软件说明
程序演示
观看下列程序演示示例:
注:新版本增加了线粒体分支节点检测功能。
功能介绍
该程序的主要功能实现如下:
- 完成细胞的分割和长宽的测量
- 完成线粒体形态参数(总长度、各段长度、段数)的统计
- 选择某一行细胞参数值可以查看其图像处理的具体过程,如图三所示。
- 自动过滤边缘的细胞
- 对于线粒体未染色或者染色不明显的细胞,仅测量其细胞形态参数
- 支持图像的批处理
软件用法
主界面介绍
程序主窗口如下:
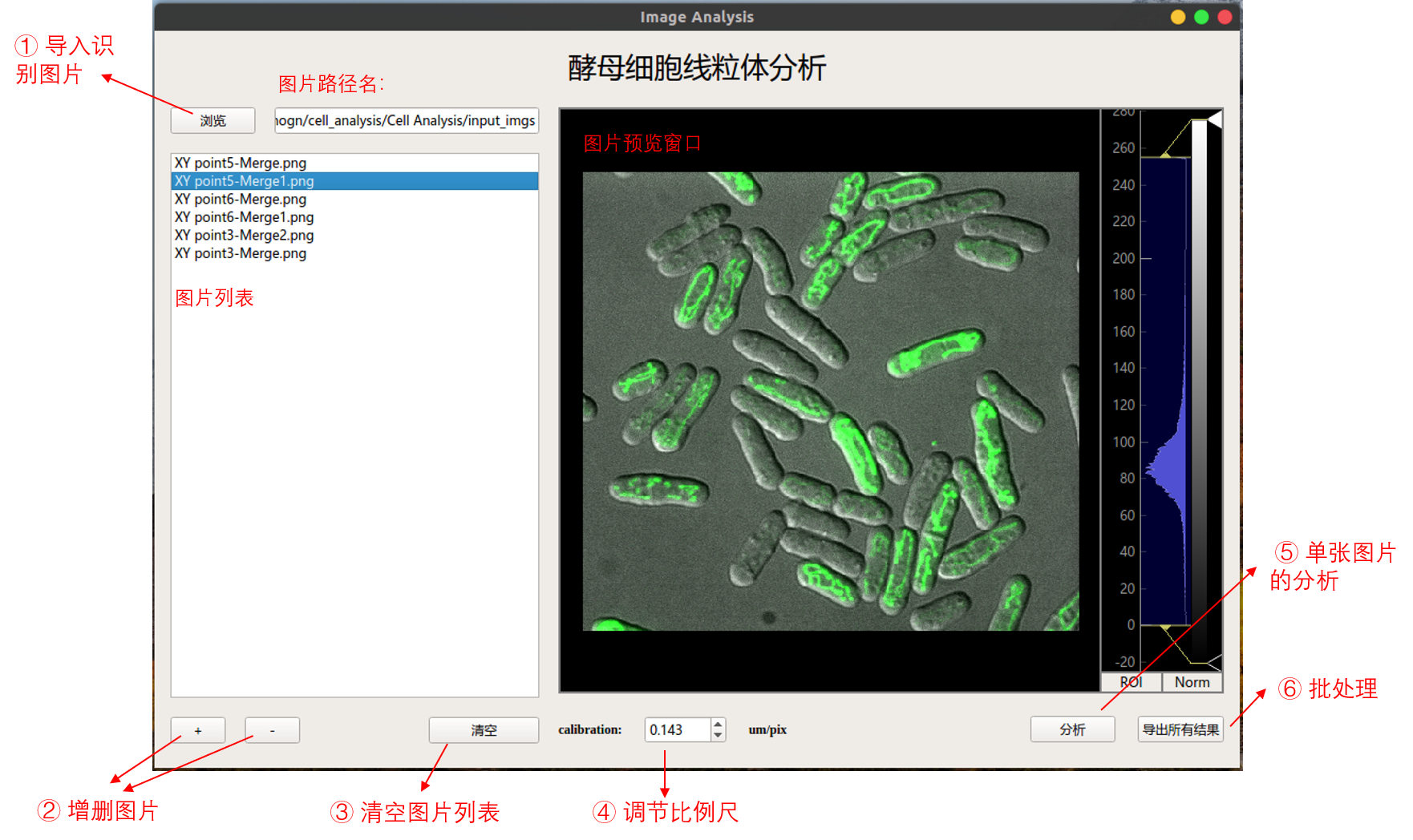
主要分为以下几个步骤:
- 点击浏览按钮打开单/多张图像
- 在左侧列表栏中选择特定图像并进行预览,通过右侧的直方图可调节图像的对比度
- 点击分析按钮,可进行当前细胞的检测以及形态参数的测量 / 点击导出所有结果按钮可以将列表中的所有图片进行检测和分析。
副界面介绍
点击主界面分析按钮弹出的结果窗口如下:
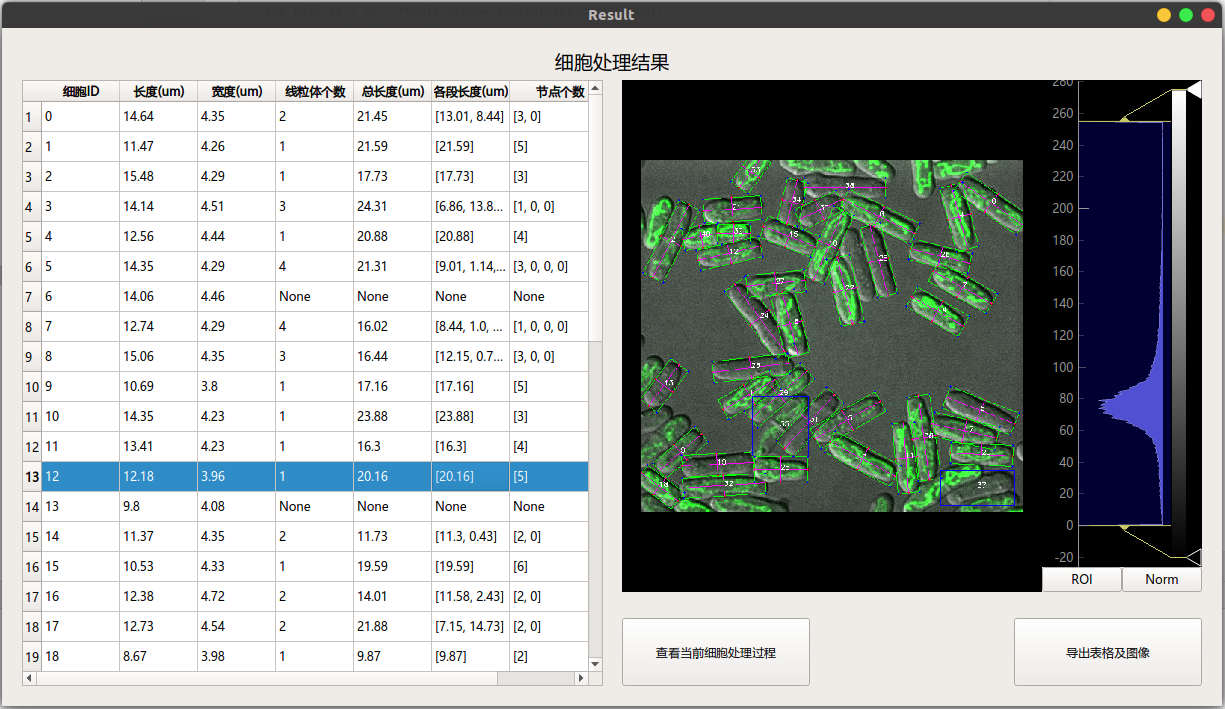
主要作如下说明:
细胞分析结果中的细胞ID 与右侧图像检测标记的数字相对应,可以根据ID快速查找对应细胞
细胞节点个数与各段长度中元素一一对应,例如,ID为0的细胞第一段线粒体长度为13.01, 对应的节点个数为3个。
选中进行预览,通过右侧的直方图可调节图像的对比度
选中细胞分析结果中的某一行数值,可以查看当前细胞的具体处理过程如下图所示。

点击导出表格和图像可以保存当前细胞的 原始图片, 检测图片, 检测结果列表
项目介绍
目标
利用上述任意图像,实现:
1. 区分各个细胞(可以利用任意通道均可)
2. 区分细胞后要测量出每一细胞的长宽度等细胞形态参数
3. 实现每一细胞内部线粒体参数(线粒体个数、各段长度、总长度)的测量。
4. 实现每一细胞内部线粒体参数(分支数量、节点数量、聚合度等)的测量。
技术路线:
按照项目的要求,可以将它分为两步走:
- 实例分割各个细胞
- 利用图像处理的形态学方法解决细胞形态参数和线粒体参数的测量
1. 实例分割
实例分割是一个具有挑战性的计算机视觉任务,需要预测的对象实例及其每像素分割掩码。这使得它混合了语义分割和目标检测。
自从 Mask R-CNN 发明以来,最先进的实例分割方法主要是 Mask R-CNN 及其变体(如 PANet、 Mask Score RCNN 等)。该算法采用先检测后分段的方法,首先执行目标检测提取每个对象实例周围的包围盒,然后在每个包围盒内执行二进制分割,分离前景(对象)和背景。
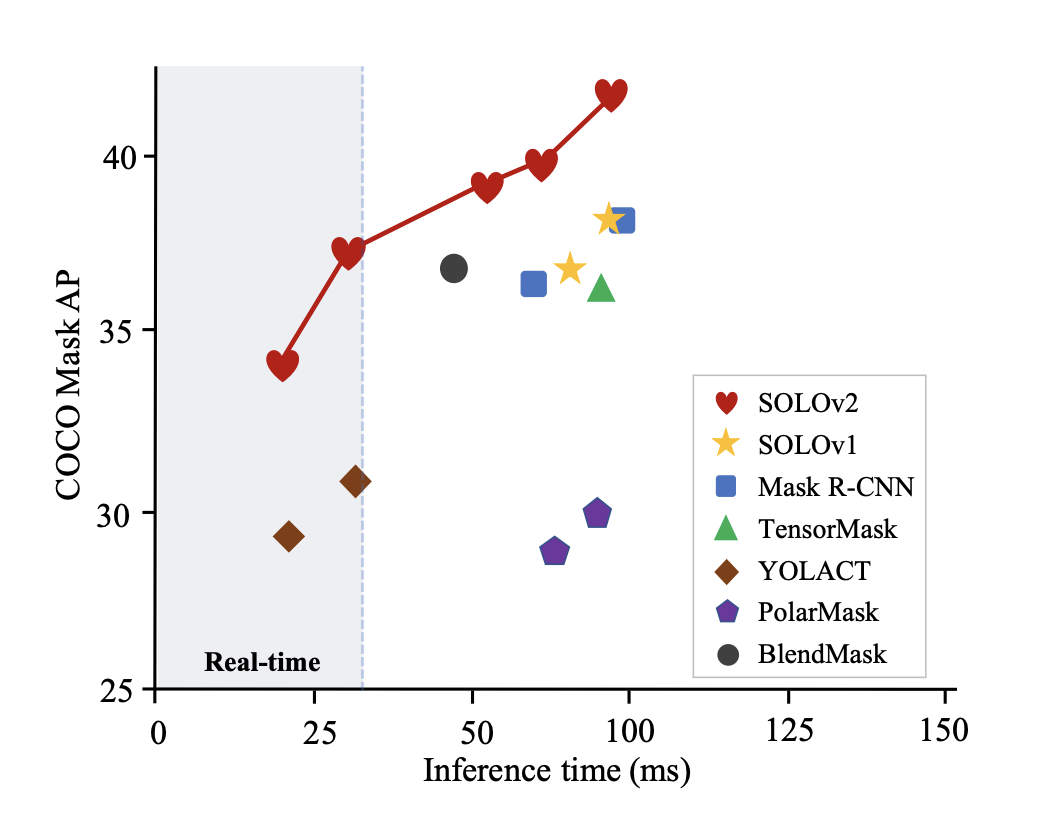
但是,Mask R-CNN 速度很慢,妨碍了许多实时应用程序的使用。此外,Mask R-CNN 预测的掩模具有固定的分辨率,因此对于具有复杂形状的大型物体没有足够的精度。随着无锚目标检测方法(如 CenterNet 和 FCOS)的进步,对单级实例分割的研究已经出现了一波研究热潮。如下图所示,这些方法中的许多比 Mask RCNN 更快更准确,但同时带来的问题是算法原理更加复杂。
在本项目中,由于细胞的形态比较单一,用最早的Mask R-CNN网络足以胜任,我先将整个项目的功能进行实现,在后续的工作完善中看情况可以将网络调整为更快的SOLO或者YOLACT,从而获得更快更准确的处理速度。
2. 图像处理形态学分析
细胞的形态学分析主要由Python的两个模块完成,分别是:python-opencv和scikit-image。具体的分析过程可参见第 4 章 细胞参数提取。
Mask R-CNN 原理
何凯明和他的团队在2018年提出的Mask R-CNN是实力分割领域最具有影响力的深度学习算法之一,近年来,我们利用Mask R-CNN框架解决了许多实际的问题。正确理解Mask R-CNN对于实际优化它的参数有着重要的意义,本文将阐述Mask R-CNN的基本原理。
基本框架
Mask-RCNN 大体框架还是 Faster-RCNN 的框架,可以说在基础特征网络之后又加入了全连接的分割子网,由原来的两个任务(分类+回归)变为了三个任务(分类+回归+分割)。Mask R-CNN 采用和Faster R-CNN相同的两个阶段:
- 第一个阶段具有相同的第一层(即RPN),扫描图像并生成提议(proposals,即有可能包含一个目标的区域);
- 第二阶段,除了预测种类和bbox回归,并添加了一个全卷积网络的分支,对每个RoI预测了对应的二值掩膜(binary mask),以说明给定像素是否是目标的一部分。所谓二进制mask,就是当像素属于目标的所有位置上时标识为1,其它位置标识为 0。
总体流程如下:
- 首先,输入一张预处理后的照片。
- 然后, 输入到一个预训练好的神经网络中(ResNet等)获得对应的特征映射(Feature map)
- 接着,对这个Feature map中的每一个点设定预定的ROI(Region of Interest),从而获得多个候选的ROI。
- 接着,对这些剩下的ROI进行ROI Align操作
- 最后,对这些ROI进行分类、回归以及MASK生成(在每个ROI里面进行FCN操作)
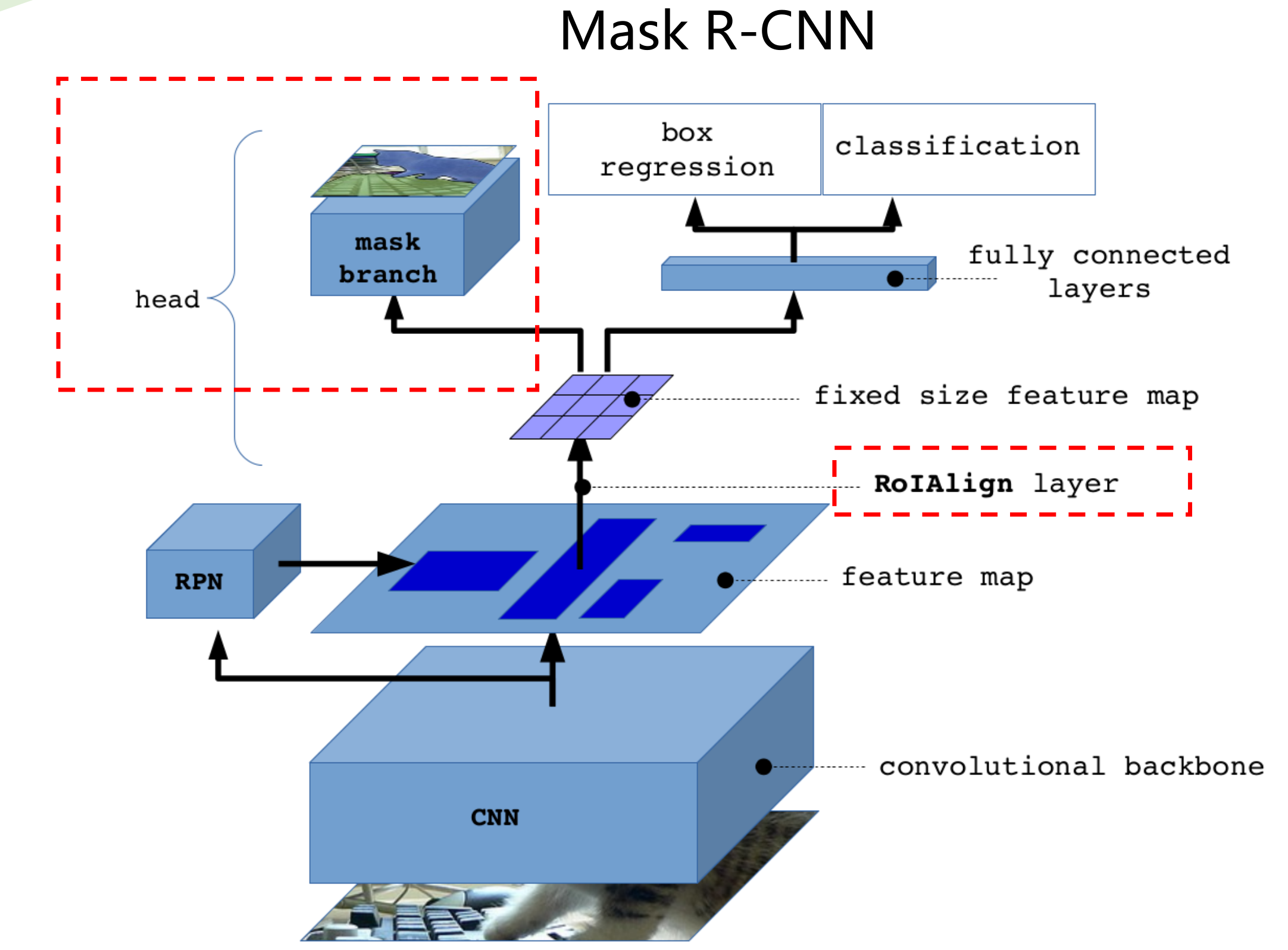
架构分解
图像预处理
为了便于训练,图像在输入网络前需要进行一定的预处理。

- Subtraction of mean:图片张量[ w, h, c ] 是输入图像需要减去所训练样本和验证样本张量的平均值,使得每个维度的平均值为0.
- Rescale: 统一输入尺寸大小为1024*1024, 若长宽不是1:1, 那么将长的一侧统一到1024,窄的一侧按比例伸缩。
- Padding: 将长度不足的一侧进行填充,填充值为0。将图片将当涉及到特征金字塔网络(FPN)时,这是必要的(在下一节中解释)。所有的填充都只在最右边缘和最底边缘完成,因此不需要改变目标的坐标,因为坐标系是从最左上角开始的。
注意: 用于生成锚点和过滤步骤的图像高度和宽度将作为缩放后的图像,而不是填充后的图像。
Feature pyramid networks (FPN) backbone
Backbone
backbone是一系列的卷积层用于提取图像的feature maps,比如可以是VGG16,VGG19,GooLeNet,ResNet50,ResNet101等,这里主要讲解的是ResNet101的结构。
ResNet(深度残差网络)实际上就是为了能够训练更加深层的网络提供了有利的思路,毕竟之前一段时间里面一直相信深度学习中网络越深得到的效果会更加的好,但是在构建了太深层之后又会使得网络退化。ResNet使用了跨层连接,使得训练更加容易。
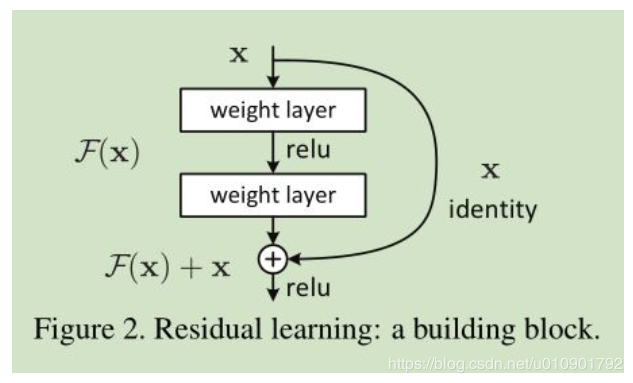
网络试图让一个block的输出为f(x) + x,其中的f(x)为残差,当网络特别深的时候残差f(x)会趋近于0,从而f(x) + x就等于了x,即实现了恒等变换,不管训练多深性能起码不会变差。
在网络中只存在两种类型的block,在构建ResNet中一直是这两种block在交替或者循环的使用,所有接下来介绍一下这两种类型的block(indetity block, conv block):
- Indetity Block
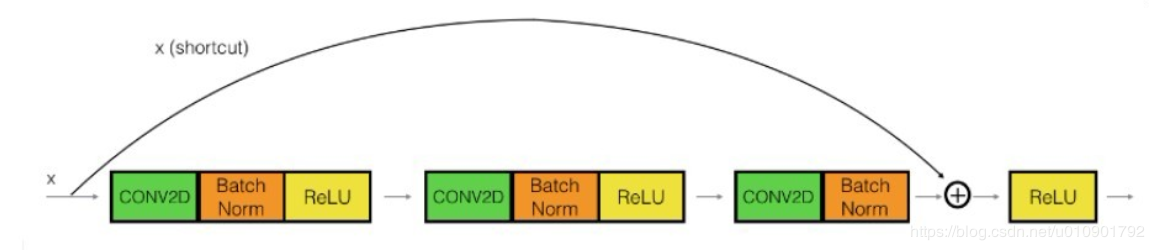
图中可以看出该block中直接把开端的x接入到第三个卷积层的输出,所以该x也被称为shortcut,相当于捷径似得。注意主路上第三个卷积层使用激活层,在相加之后才进行了ReLU的激活。
- Conv Block
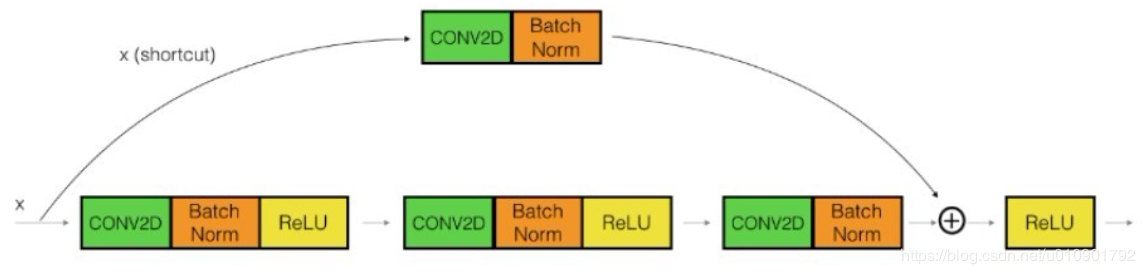
与identity block其实是差不多的,只是在shortcut上加了一个卷积层再进行相加。注意主路上的第三个卷积层和shortcut上的卷积层都没激活,而是先相加再进行激活的。
其实在作者的代码中,主路中的第一个和第三个卷积都是1*1的卷积(改变的只有feature maps的通道大小,不改变长和宽),为了降维从而实现卷积运算的加速;注意需要保持shortcut和主路最后一个卷积层的channel要相同才能够进行相加。下面展示Res-net101的完整框架:
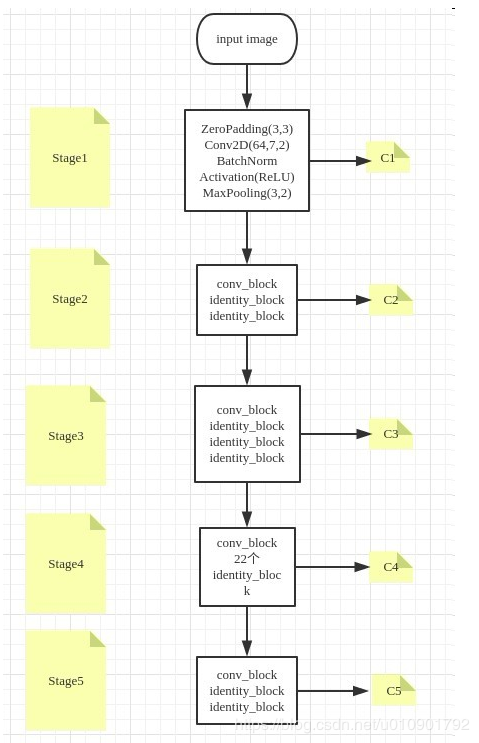
从图中可以得知ResNet分为了5个stage,C1-C5分别为每个Stage的输出,这些输出在后面的FPN中会使用到。你可以数数,看看是不是总共101层,数的时候除去BatchNorm层。注:stage4中是由一个conv_block和22个identity_block,如果要改成ResNet50网络的话只需要调整为5个identity_block.
Feature Pyramid Map
概念
FPN的提出是为了实现更好的feature maps融合,一般的网络都是直接使用最后一层的feature maps,虽然最后一层的feature maps 语义强,但是位置和分辨率都比较低,容易检测不到比较小的物体。FPN的功能就是融合了底层到高层的feature maps ,从而充分的利用了提取到的各个阶段的特征(ResNet中的C2-C5 )。简单来说,就是把底层的特征和高层的特征进行融合,便于细致检测。
FPN是为了自然地利用CNN层级特征的金字塔形式,同时生成在所有尺度上都具有强语义信息的特征金字塔。所以FPN的结构设计了top-down结构和横向连接,以此融合具有高分辨率的浅层layer和具有丰富语义信息的深层layer。*这样就实现了*从单尺度的单张输入图像,快速构建在所有尺度上都具有强语义信息的特征金字塔,同时不产生明显的代价。
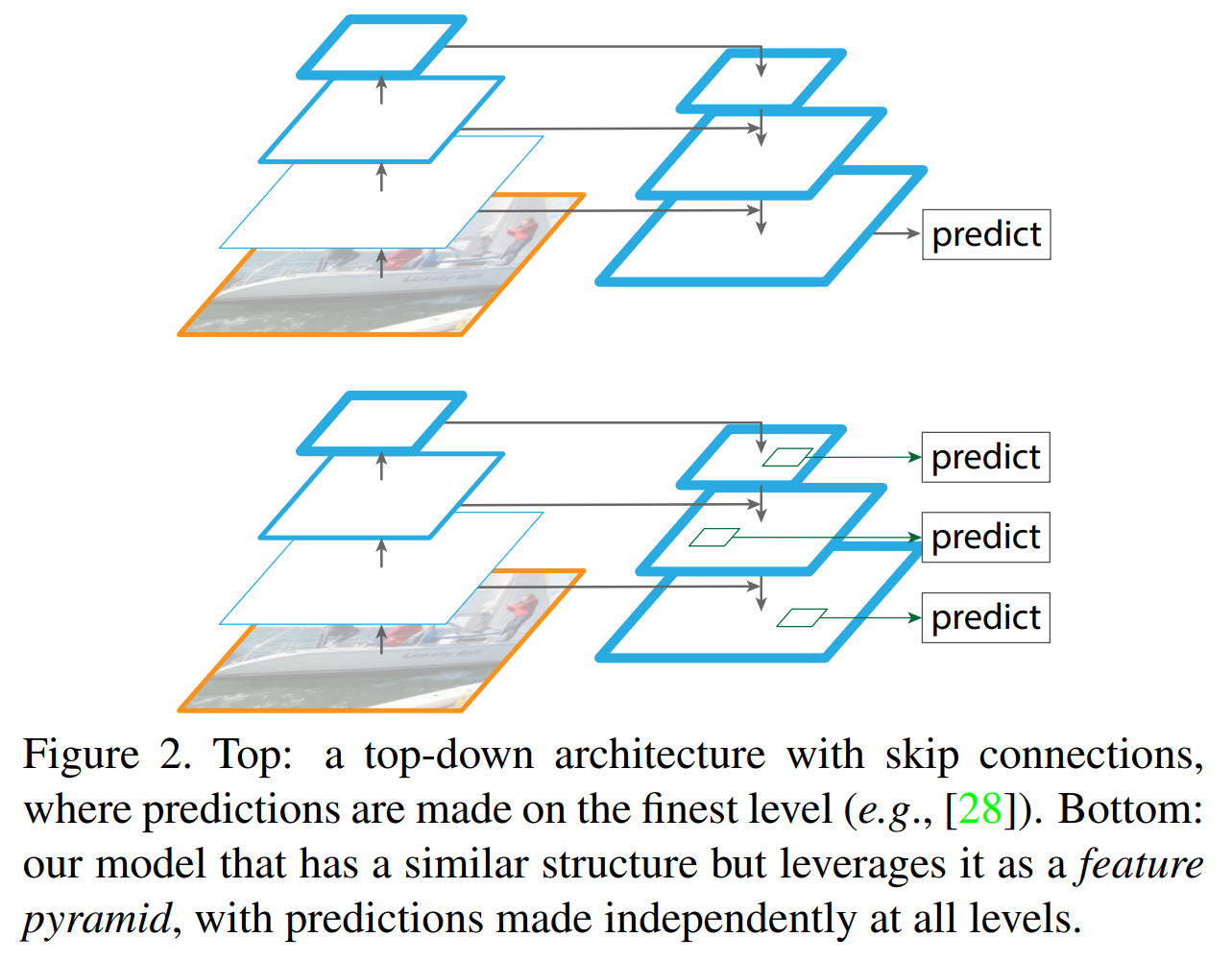
如下图所示: Top: 一个带有skip connection的网络结构在预测的时候是在finest level(自顶向下的最后一层)进行的,简单讲就是经过多次上采样并融合特征到最后一步,拿最后一步生成的特征做预测。Bottom: FPN网络结构和上面的类似,区别在于预测是在每一层中独立进行的。后面的实验证明finest level的效果不如FPN好,原因在于FPN网络是一个窗口大小固定的滑动窗口检测器,因此在金字塔的不同层滑动可以增加其对尺度变化的鲁棒性。另外虽然finest level有更多的anchor,但仍然效果不如FPN好,说明增加anchor的数量并不能有效提高准确率。
特征融合图
特征金字塔提取图像特征可以分为两个部分: 自下而上(down-to-top pathway)和自上而下(top-to-down pathway)两条路径,或者将自下而上路径称为编码过程,自上而下路径称为解码过程。
Down-to-top pathway
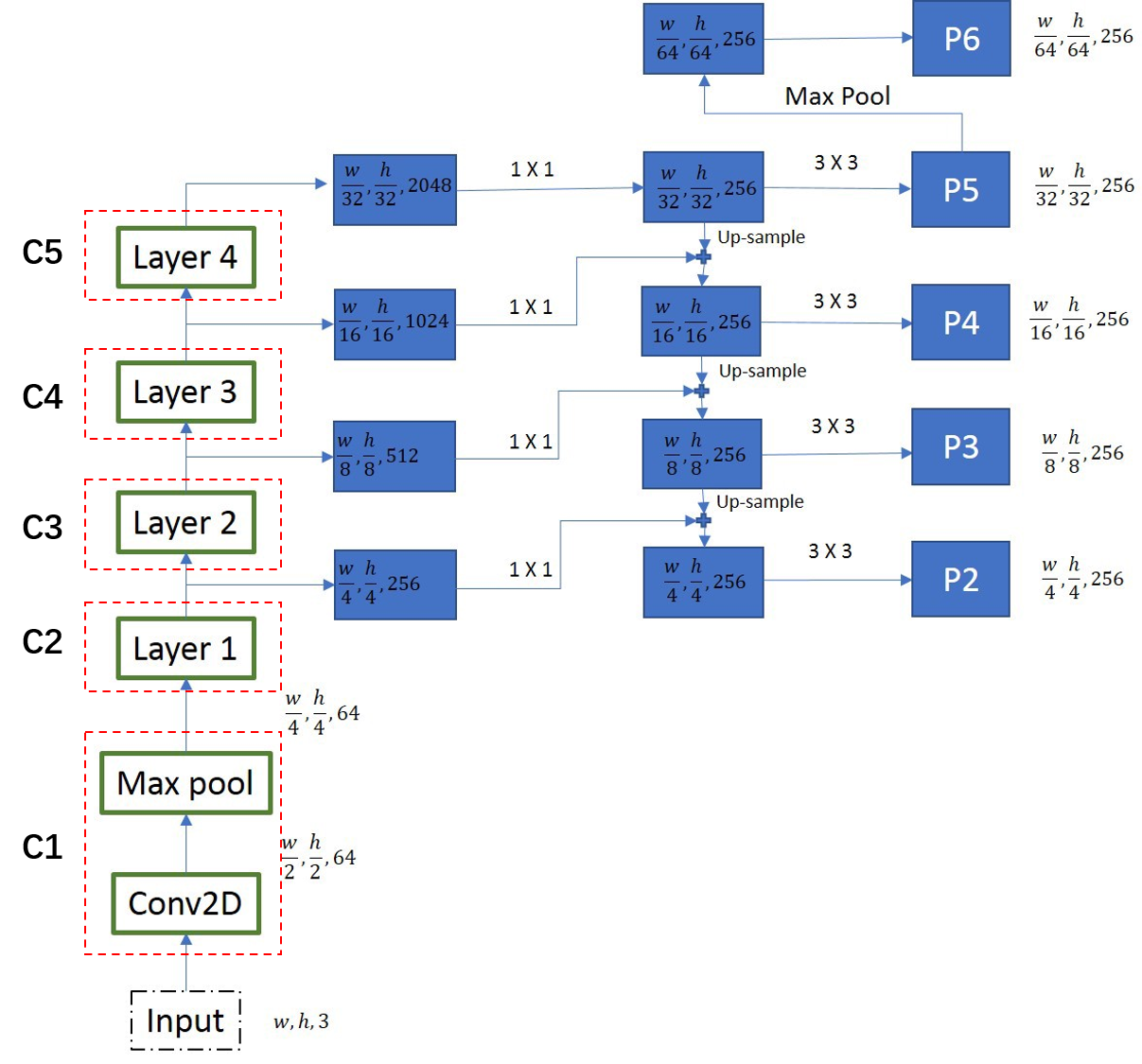
CNN的前馈计算就是自下而上的路径,特征图经过卷积核计算,通常是越变越小的,也有一些特征层的输出和原来大小一样,称为“相同网络阶段”(same network stage )。对于本文的特征金字塔,作者为每个阶段定义一个金字塔级别, 然后选择每个阶段的最后一层的输出作为特征图的参考集。 这种选择是很自然的,因为每个阶段的最深层应该具有最强的特征。具体来说,对于ResNets,作者使用了每个阶段的最后一个残差结构的特征激活输出。将这些残差模块输出表示为{C2, C3, C4, C5},对应于conv2,conv3,conv4和conv5的输出,并且注意它们相对于输入图像具有{4, 8, 16, 32}像素的步长(也就是感受野)。考虑到内存占用,没有将conv1包含在金字塔中。
Top-to-down pathway
自上而下的路径(the top-down pathway )是如何去结合低层高分辨率的特征呢?方法就是,把更抽象,语义更强的高层特征图进行上取样,然后把该特征横向连接(lateral connections )至前一层特征,因此高层特征得到加强。值得注意的是,横向连接的两层特征在空间尺寸上要相同。这样做应该主要是为了利用底层的定位细节信息。
下图显示连接细节。把高层特征做2倍上采样(最邻近上采样法,可以参考反卷积),然后将其和对应的前一层特征结合(前一层要经过1 * 1的卷积核才能用,目的是改变channels,应该是要和后一层的channels相同),结合方式就是做像素间的加法。重复迭代该过程,直至生成最精细的特征图。迭代开始阶段,作者在C5层后面加了一个1 * 1的卷积核来产生最粗略的特征图,最后,作者用3 * 3的卷积核去处理已经融合的特征图(为了消除上采样的混叠效应),以生成最后需要的特征图。为了后面的应用能够在所有层级共享分类层,这里作者固定了3*3卷积后的输出通道为d,这里设为256.因此所有额外的卷积层(比如P2)具有256通道输出。这些额外层没有用非线性。
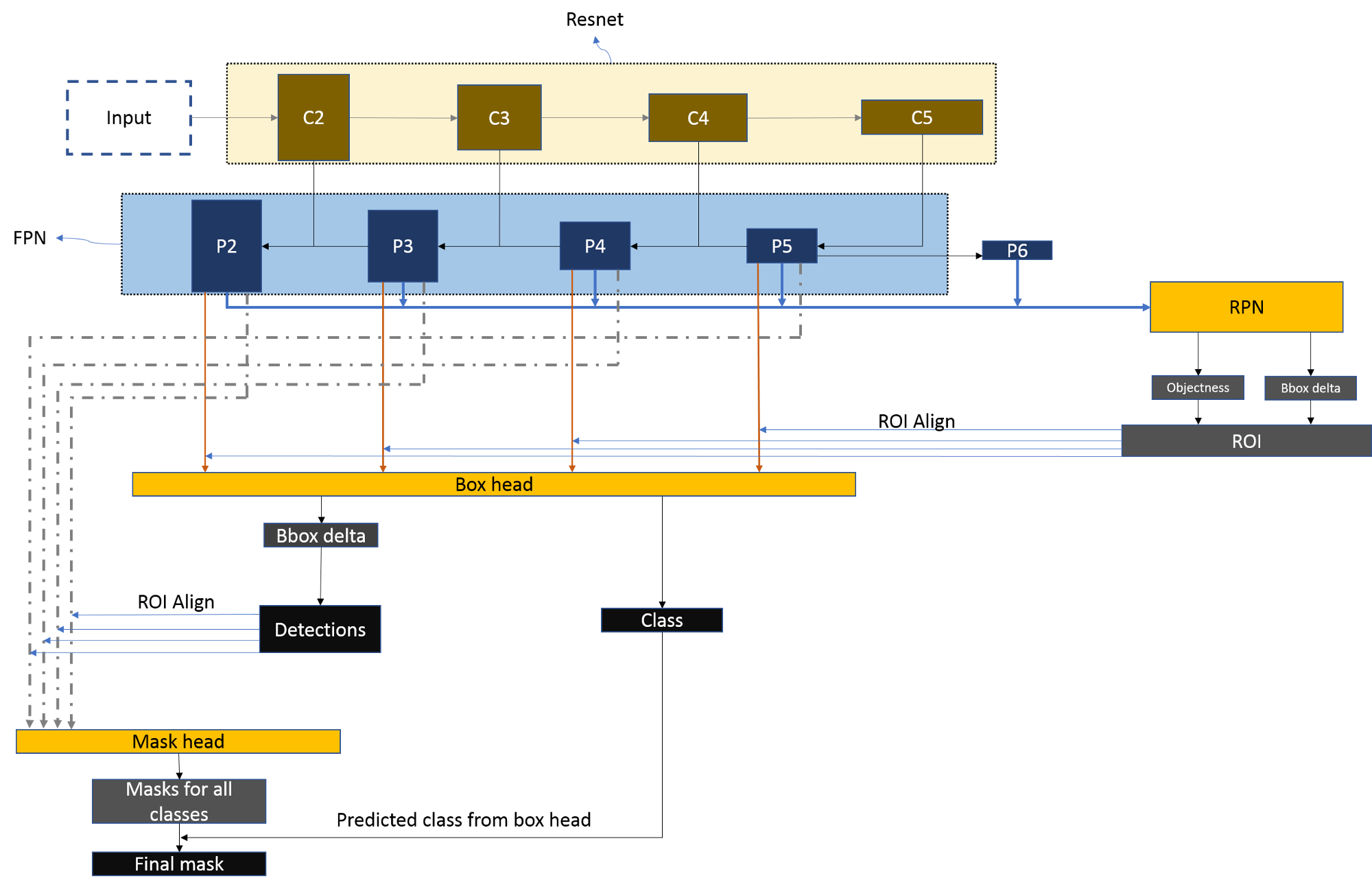
{C2, C3, C4, C5}层对应的融合特征层为{P2, P3, P4, P5},对应的层空间尺寸是相通的。
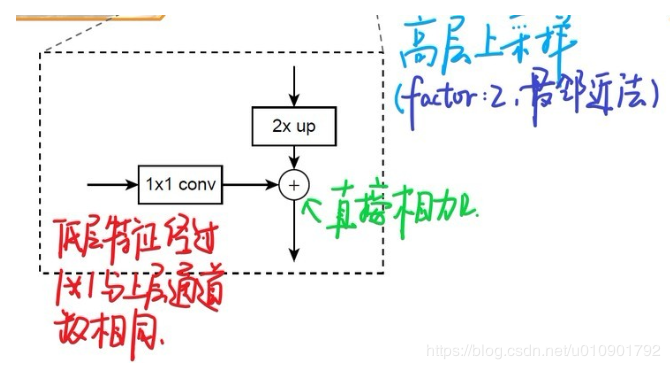
从图中可以看出+的意义为:左边的底层特征层通过1×1的卷积得到与上一层特征层相同的通道数;上层的特征层通过上采样得到与下一层特征层一样的长和宽再进行相加,从而得到了一个融合好的新的特征层。举个例子说就是:C4层经过1×1卷积得到与P5相同的通道,P5经过上采样后得到与C4相同的长和宽,最终两者进行相加,得到了融合层P4,其他的以此类推。
注:P2-P5是将来用于预测物体的bbox,box-regression,mask的,而P2-P6是用于训练RPN的,即P6只用于RPN网络中。
输出
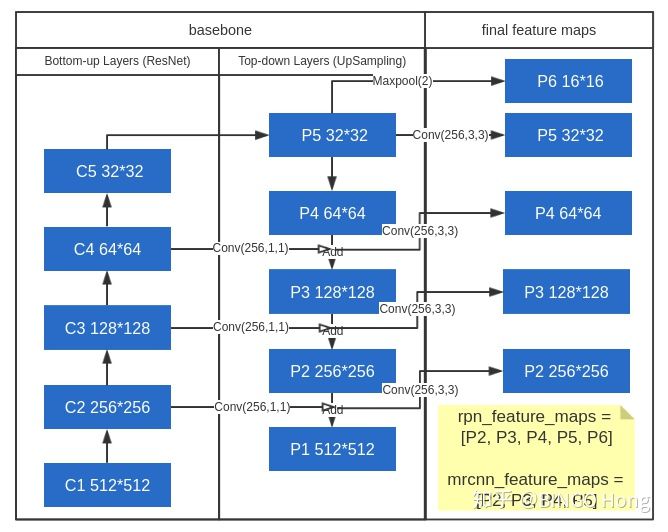
经过高层特征与底层特征的融合之后,每层进行特征输出,分别得到[P2, P3, P4, P5, P6]五层。其中RPN网络将用到[P2, P3, P4, P5, P6], 而目标检测网络部分用到[P2, P3, P4, P5],也就是说,P6 16×16的特征张量只用于RPN(感兴趣区域生成网络中),用途后面具体再聊。
- rpn_feature_maps: [P2, P3, P4, P5, P6]
- mrcnn_feature_maps: [P2, P3, P4, P5]
| 特征层 | 形状 |
|---|---|
| P2 | 256*256 @ 256 |
| P3 | 128*128 @ 256 |
| P4 | 64*64 @ 256 |
| P5 | 32*32 @ 256 |
| P6* | 16*16 @ 256 |
Proposal Region Of Interest
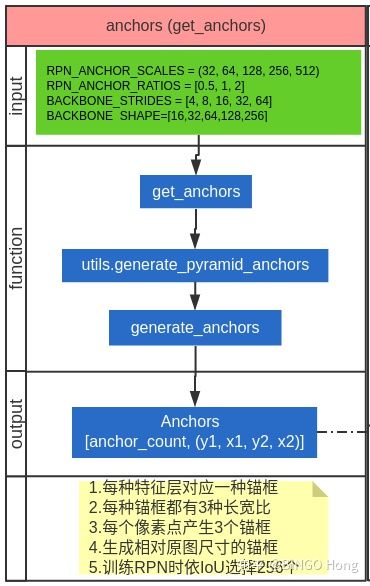
第二部分主要介绍感兴趣区域(ROI)的生成规则,他主要有三个部分组成:锚框(anchor)生成、区域提议网络(Region Proposal Network,RPN)、区域生成网络(Proposal Layer)。如下图所示,最左侧的anchors是生成锚框流程,中间的build_rpn_model->rpn_graph是构建RPN网络流程,最右侧的ProposalLayer是筛选ROIs的生成建议框流程。
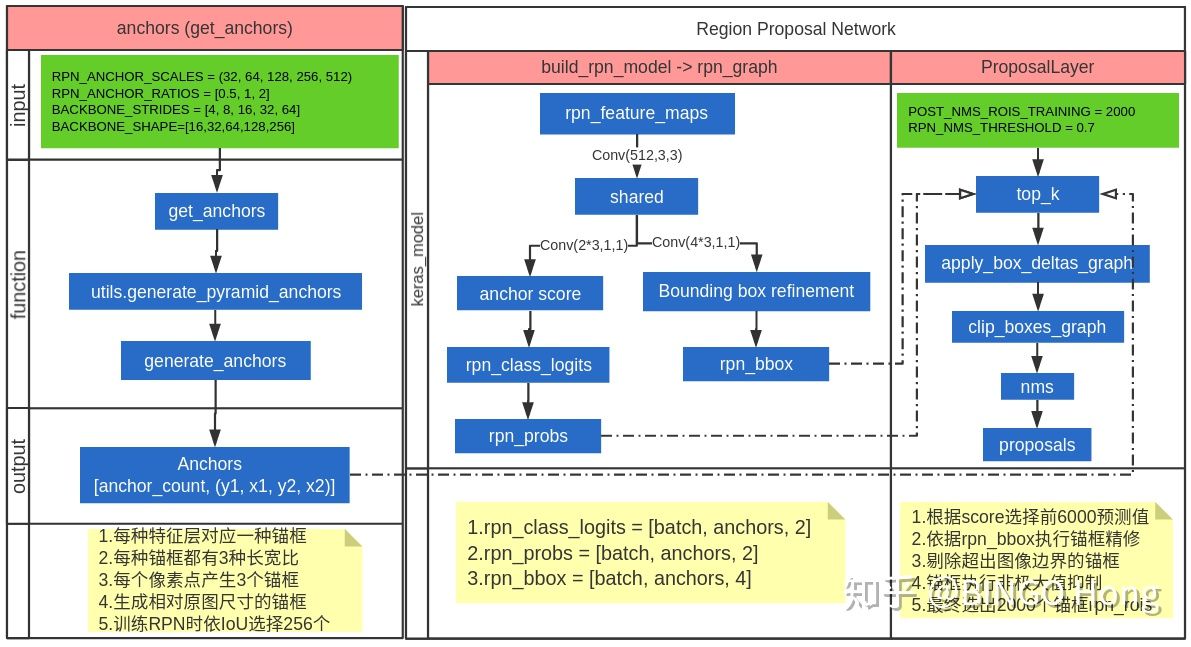
锚框生成
首先,需要理解锚框的概念,在这篇博文中对锚框有着很好的说明。笼统的说,P2~P6的特征图相当于将原来1024×1024的图像进行了分块,将每一块都压缩成一个像素点,然后,以该像素点为中心进行框选周边区域,为了检测多种形状的物体,同一个像素区域利用多个锚框进行框选。具体参数如下:
配置参数
- RPN_ANCHOR_SCALES是anchor尺寸,分别为 (32, 64, 128, 256, 512),对应rpn_feature_maps的[P2, P3, P4, P5, P6],分辨率依次为[256,128,64,32,16],也就是说底层高分辨率特征去检测较小的目标,顶层低分辨率特征图用于去检测较大的目标。
- RPN_ANCHOR_RATIOS是锚框的长宽比,对应每一种尺寸的锚框取[0.5, 1, 2],3种长宽比
- BACKBONE_STRIDES 是特征图的降采样倍数,取[4, 8, 16, 32, 64]
- BACKBONE_SHAPE是特征图分辨率,为[16,32,64,128,256]
generate_anchors是具体的为每一特征层生成anchor的函数,generate_pyramid_anchors用于拼接不同scale的anchor,最终得到anchors的shape为[anchor_count, (y1, x1, y2, x2)],此时计算的anchor_count = (256×256 + 128×128 + 64×64 + 32×32 + 16×16)3 = 261888。数量如此多的锚框不可能全部用于预测,所以有了后续的proposallayer进行筛选。
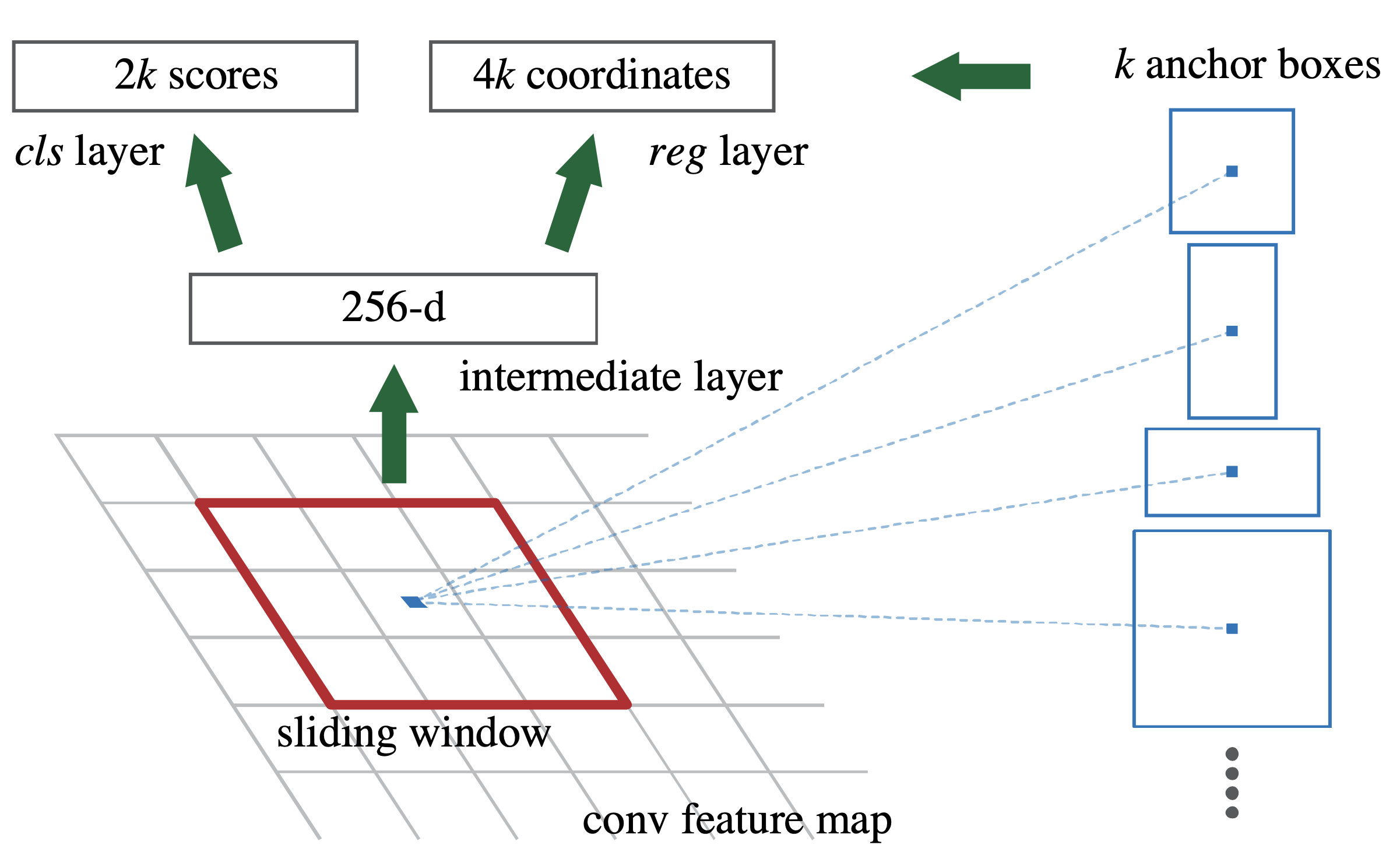
RPN 网络
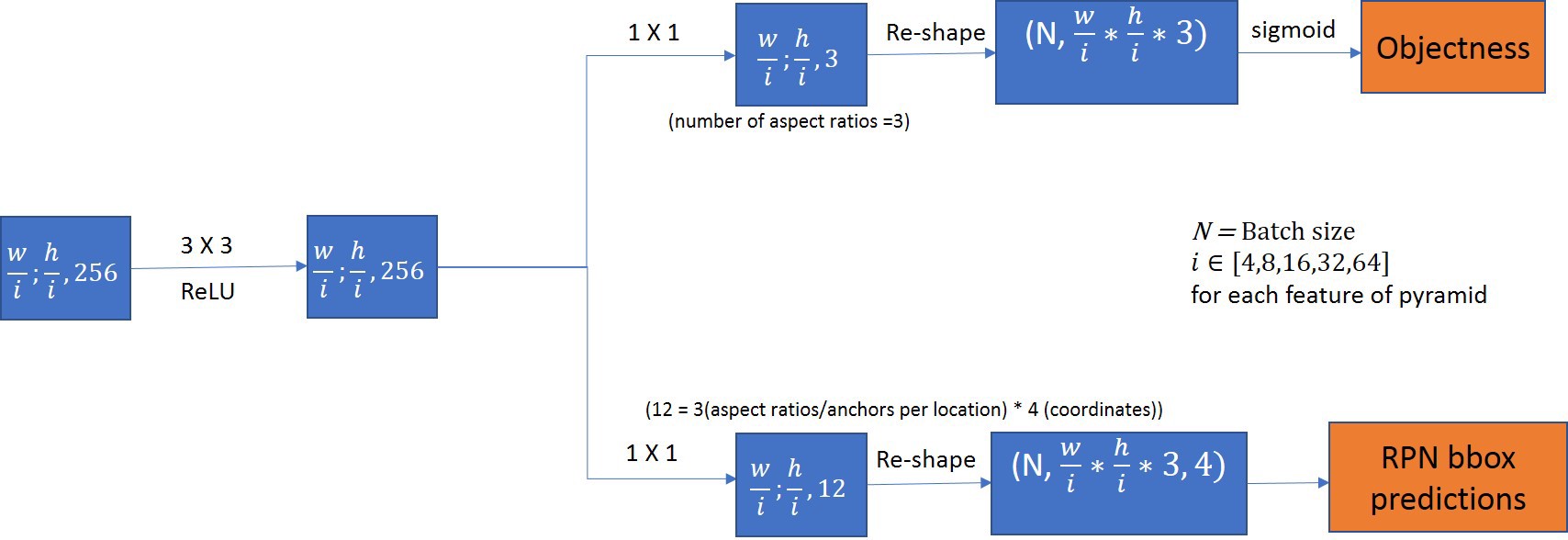
FPN输出的特征映射张量[P2, P3, P4, P5, P6]将用于RPN网络,P[?]代表的张量形状各不相同,因此需要分开处理,RPN网络的结构如下图所示,以P2为例:
P2的大小为256×256@256,经过一次(512,3,3)的卷积操作,形状变为[256, 256, 512]
然后分成两路:一路用于目标概率的判断,另一路用于目标框坐标的推断
Objectness:
- 利用1×1的全卷积进行特征维度的压缩。由于每个目标框的anchors数目为3,并且需要分别给出前景和背景的概率值,因此,特征维度需要压缩为2×anchors_per_location个,也就是6个,输出形状为[256, 256, 6]
- 将[256, 256, 6]的输出重塑为[ 256×256×3,2],其中第一维度代表anchors的总个数,第二维度代表前景和背景的概率值,最后,通过softmax函数,将概率值转换为类别(One-hot编码)
RPN bbox predictions:
- 利用1×1的全卷积进行特征维度的压缩,由于每个目标框的anchors数目为3,一个box的定位需要4个坐标值(x1,y1,x2,y2),所以一个像素点所需要的的向量长度为3×4=12,然后,将anchors数目在空间维度进行融合,得到256×256×3个anchor,输出的张量形状为[256×256×3, 4]
其他特征张量的处理方式上同,作者在这里采用了循环处理的方式,并且共享了RPN的权重。
注意:以上均是以一张图片的处理过程为例,在实际的操作中,还有batchsize一个维度加在张量的最前面。
最终输出形式:
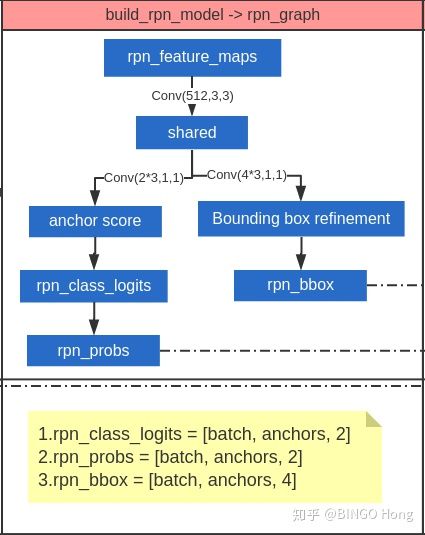
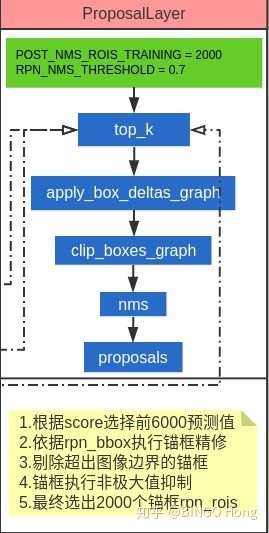
Proposal Layer
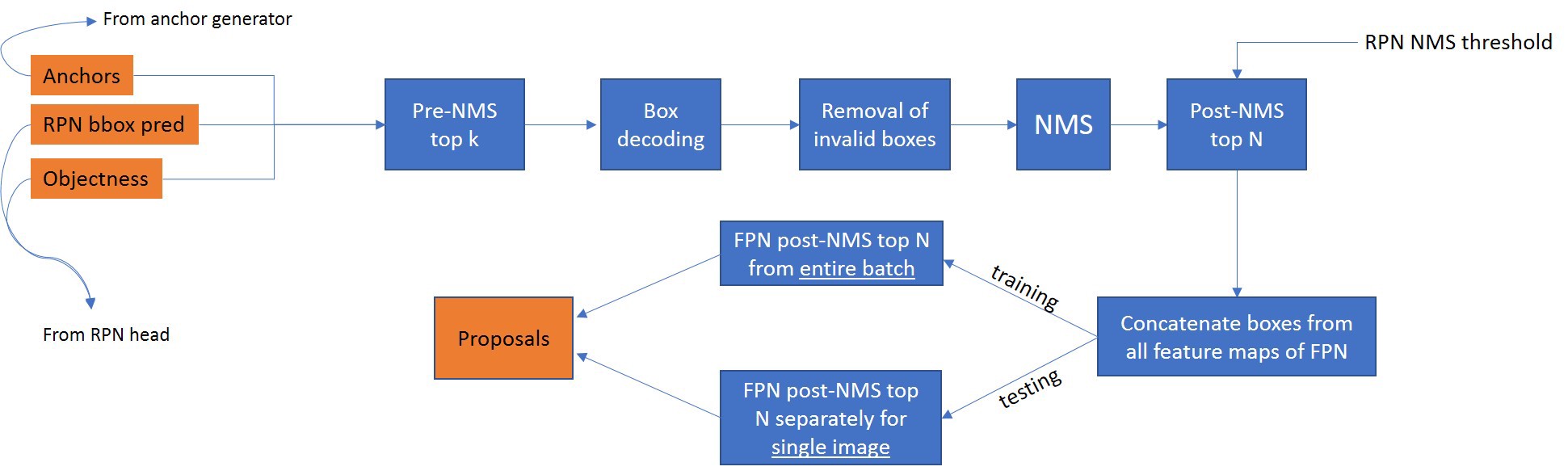
Proposal Layer的输入来自于前两部分:Anchors生成和RPN输出。在Proposal Layer中,并没有需要进行训练的参数,只是利用算法进行anchors的选择与修正工作。
- proposallayer需要3个输入参数依次为[rpn_class, rpn_bbox, anchors],自定义层内部表示为[scores, deltas, anchors],其中rpn_class和rpn_bbox为2.2预测分类和回归框偏移,2.1生成的anchors,各有261888个。在训练mrcnn时,一张图片不可能用这么多rois,因此要进一步筛选。
- Pre-NMS top k:用于根据rpn_class概率(也可以理解为rois得分)选取至多前6000个rois的索引,然后再根据索引选择出相应的属于top-6000的[scores, deltas, anchors]。tf.gather用于根据y索引选择x值。
- Box decoding:用于根据deltas对anchors进行精修
- removal of invalid boxes用于将超出图片范围的anchors进行剔除,这里由于回归框是归一化在[0,1]区间内,所以通过clip进行限定。
- nms执行非极大值抑制,根据IoU阈值选择出2000个rois,如果选择的rois不足2000,则用0进行pad填充。
- 最终返回的proposals赋值给rpn_rois,作为rpn网络提供的建议区,注入后续FPN heads进行分类、目标框和像素分割的检测。(图中Concatenate boxes from all feature maps of FPN应该是pytorch版本的实现方法,在本结构中,在rpn网络输出的时候就已经将各特征层融合在一起了。)
Network Heads
利用rpn_feature_map和anchors提取出rois,rois和maskrcnn_feature_map一起输入到网络的第三部分:Network Heads。
RoIAlign layer
RoIAlign的作用是根据rois的坐标,在feature_map中将对应的区域框选出来,然后利用插值法则,生成固定尺寸的张量输入到分类网络和分割网络。这里有两个概念需要理清楚。
RoIAlign的概念
离散数据和连续数据不一样,由于rois中的坐标值是离散的,而feature_map中的值是离散的,所以在进行区域截取的时候必然会发生区域的错位现象,为了保证区域的对齐,作者在这里采用了一种叫做RoIAlign的算法,利用二次插值,计算出rois坐标位置的插值,然后进行点的计算与下采样,这样得到的fixed_size_feature_map与rois标注的区域差别最小。
4.1.1 feature_map和rois 
4.1.2 fast_rcnn采用的区域选择方式 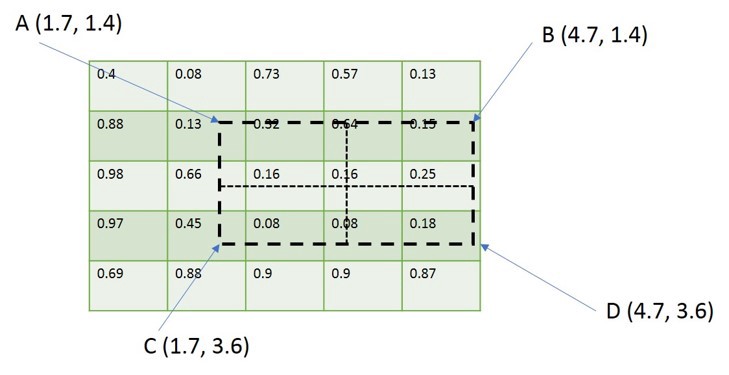
4.1.3 mask_rcnn采用的区域对齐方式 feature_map的选择
从2.3节中有提到,mrcnn_feature_maps是[P2, P3, P4, P5]的组合向量,每一个输出的特征图大小各不相同,每一个rois需要对应其中的1张特征图进行截取,所以对于特征图的选择需要有一定的规则。这里作者定义了一个系数Pk,其定义为:
其中, 代表的是基准值,设置为5,代表P5层的输出, 代表的是roi的长度和宽度, 224是ImageNet的标准输入大小。例如,若, 那么 的取值为4,rois需要截取P4输出的特征值
结构
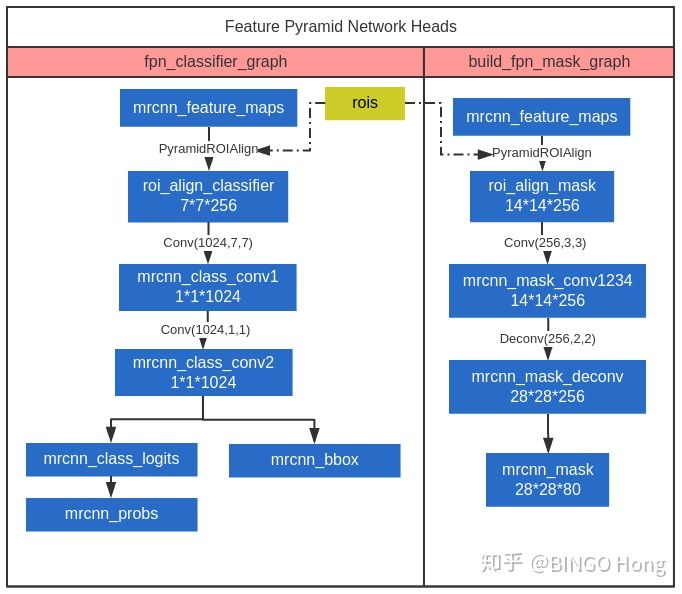
整体FPN heads分为两个分支,一是用于分类和回归目标框偏移的fpn_classifier_graph,一是用于像素分割的build_fpn_mask_graph,两者都是将rois在对应mrcnn_feature_maps特征层进行roialign特征提取,然后再经过各自的卷积操作预测最终结果。
PyramidROIAlign层不展开讨论,可认为将pool_size划分的77区域(对于mask_pool_size则为1414)取若干采样点后,进行双线性插值得到f(x,y),这个版本的代码中取采样点为1。
keras.layers.TimeDistributed作为封装器可将一个层应用于输入的每个时间片,要求输入的第一个维度为时间步。该封装器在搭建需要独立连接结构时用到,例如mask rcnn heads结构,进行类别判断、box框回归和mask划分时,需要对num_rois个感兴趣区域ROIs进行分类回归分割处理,每一个区域的处理是相对独立的,此时等价于时间步为num_rois,
fpn_classifier_graph
首先利用PyramidROIAlign提取rois区域的特征,再利用TimeDistributed封装器针对num_rois依次进行77->11卷积操作,再分出两个次分支,分别用于预测分类和回归框。解释如下:
- PyramidROIAlign用于提取rois区域特征,输出维度为[batch, num_boxes, 7,7,256]
- TimeDistributed封装器针对num_rois依次进行77->11卷积操作,维度变化为[batch, num_boxes, 1,1,1024],此时num_rois相当于时间步独立操作。
- 最后输出内容为[mrcnn_class_logits, mrcnn_probs, mrcnn_bbox],其中mrcnn_probs是预测概率,mrcnn_bbox是预测目标框偏移量
build_fpn_mask_graph
首先利用PyramidROIAlign提取rois区域的特征,再利用TimeDistributed封装器针对num_rois依次进行33->33->33->33卷积操作,再经过2*2的转置卷积操作,得到像素分割结果。解释如下:
- PyramidROIAlign用于提取rois区域特征,输出维度为[batch, num_boxes, 14,14,256]
- 4层常规卷积层整合特征
- 最终输出Masks做为分割结果,维度为[batch, num_rois, 28, 28, 80],这里为每一类实例都提供一个channel,原论文的观点是"避免了不同实例间的种间竞争"。
Mask R-CNN 实现
Mask R-CNN 核心模型代码在 GitHub开源, 该项目在Matterport/Mask_RCNN的基础上进行开发。可直接将该项目克隆到本地。
xxxxxxxxxx$ git clone https://github.com/matterport/Mask_RCNN.git数据集制作

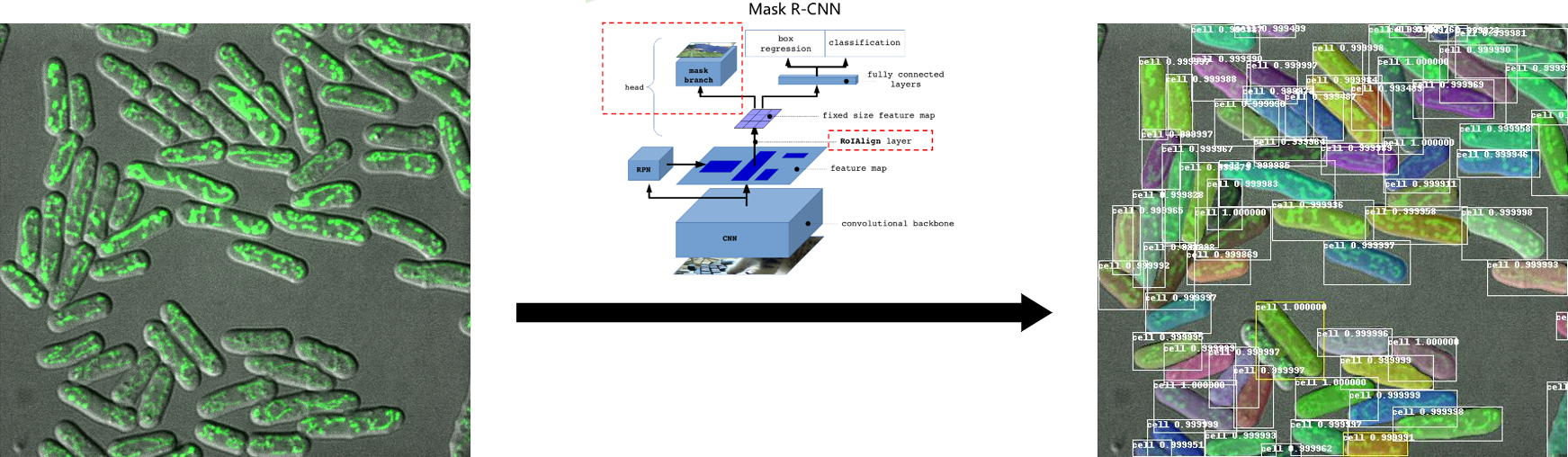
上图是利用Mask-RCNN网络以及已经训练好的模型进行人物分割的一个演示,分割的结果不仅将每个人物进行的框选,并且对人物进行了语义的分割。如果将以上图片中的人物换做是显微镜下的细胞,经过网络同样形式的训练,可想而知,也是可以达到同样效果的。Mask-RCNN的核心代码部分已经实现,现在只需要将当前测试的人物数据集换成细胞数据集,下面开始介绍细胞数据集标注的具体方式。
相应软件
为了方便不同平台的标注,标注软件采用的是:VGG Image Annotator (VIA),这是一个网页版的应用,无需安装,简洁,轻量。
图像标记
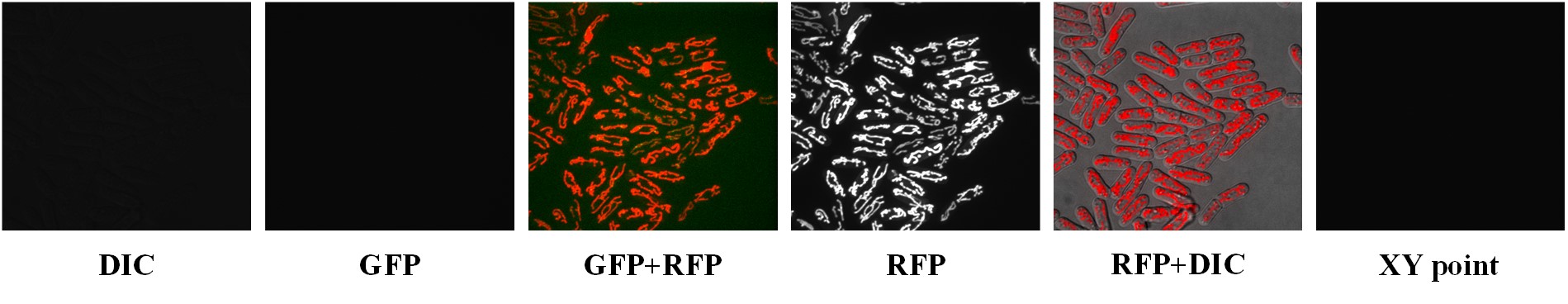
网络输入图像将采用RFP+DIC处理后的JPG图,其中细胞的轮廓以及线粒体的形态都比较清晰,有利于以后细胞形态以及线粒体参数的提取。由于细胞标注的工作量较大,所以最好可以将现有的数据图片分成几组分别进行标注。
由于目前没有获得RFP+DIC的完整数据集,所以下面将以matlab处理的伪彩色图数据集加以演示:
导入图像集
首先通过Remove健将演示图片删除,然后点击Add Files将图片集导入到VIA中。为了便于右侧图片的精细标注,利用Ctrl+鼠标滚轮将图片放大到合适的大小。
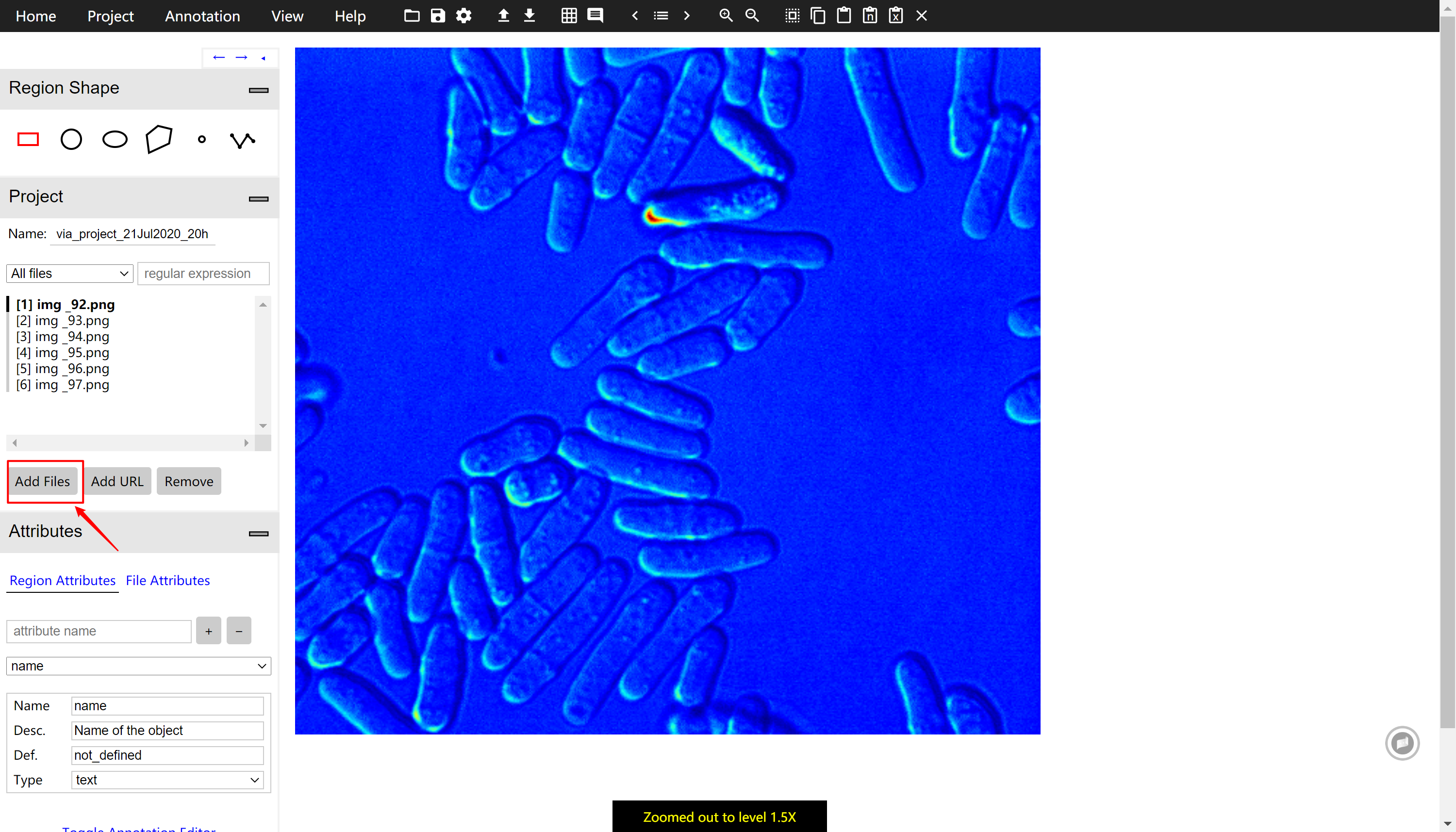
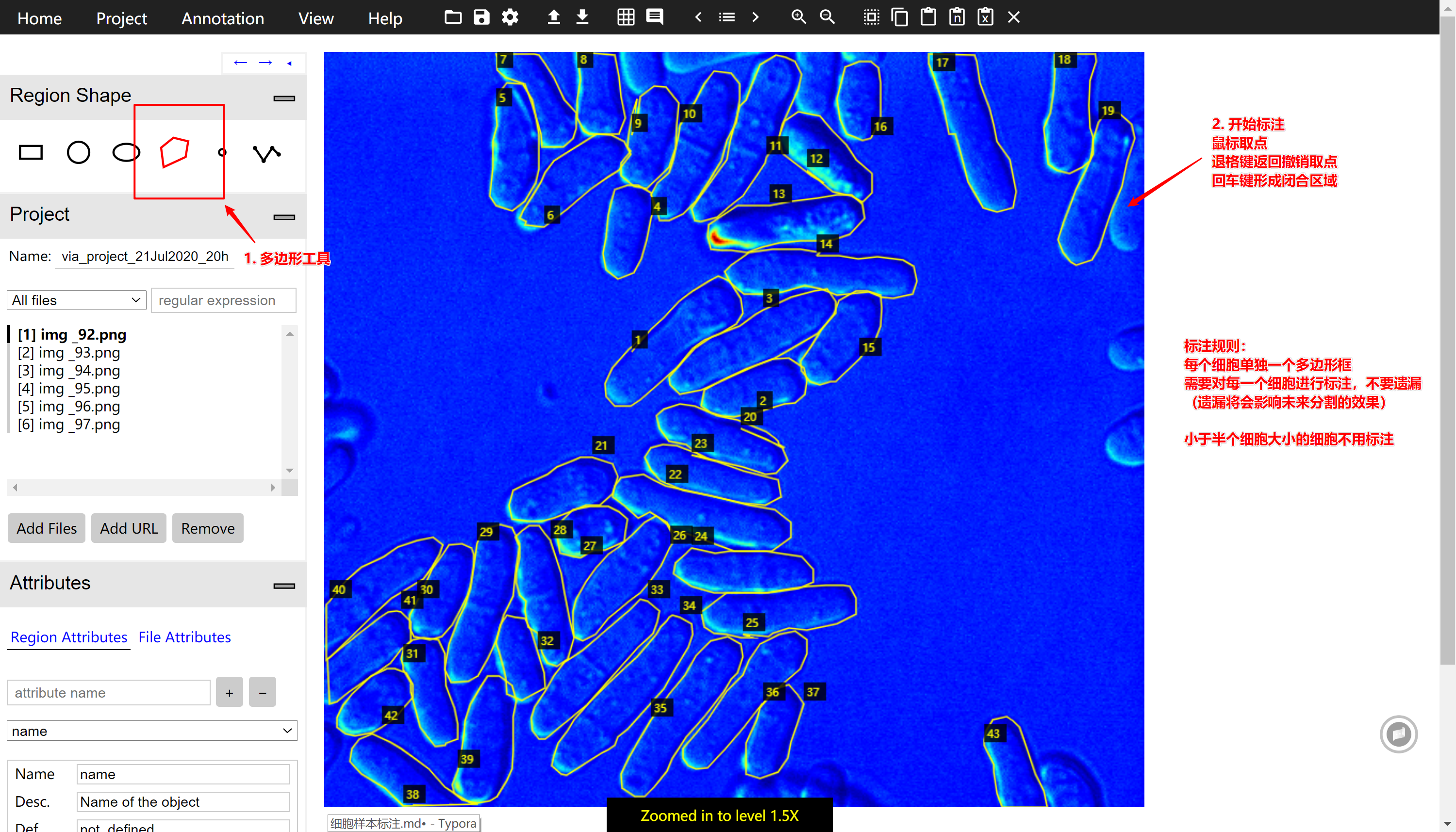
开始多边形标注
首先,在Region Shape菜单中选中多边形状工具,然后进行细胞样本的标注,如下图所示。
xxxxxxxxxx* 利用鼠标,在每个细胞的边缘进行描点,利用`Backspace`撤销当前点,利用`Enter`键来确定当前区域,形成完整的闭合区域,然后进行下一个细胞的标注。
标注的规则:
- 将图片放大一点,尽量贴近细胞的外边沿描框。
- 各个细胞多边形框之间避免重合,若遇到两个细胞重叠的区域,将重叠的区域归类给在上层的细胞。
- 标注完后检查有无未被标注的细胞,避免遗漏。标注遗漏将会影响未来细胞实例分割的准确性。(在边缘区域只有不到一半的细胞也需要标注(图片中写错了)。
经过试验,标注这样一幅图像的时间大概在10-15分钟左右。
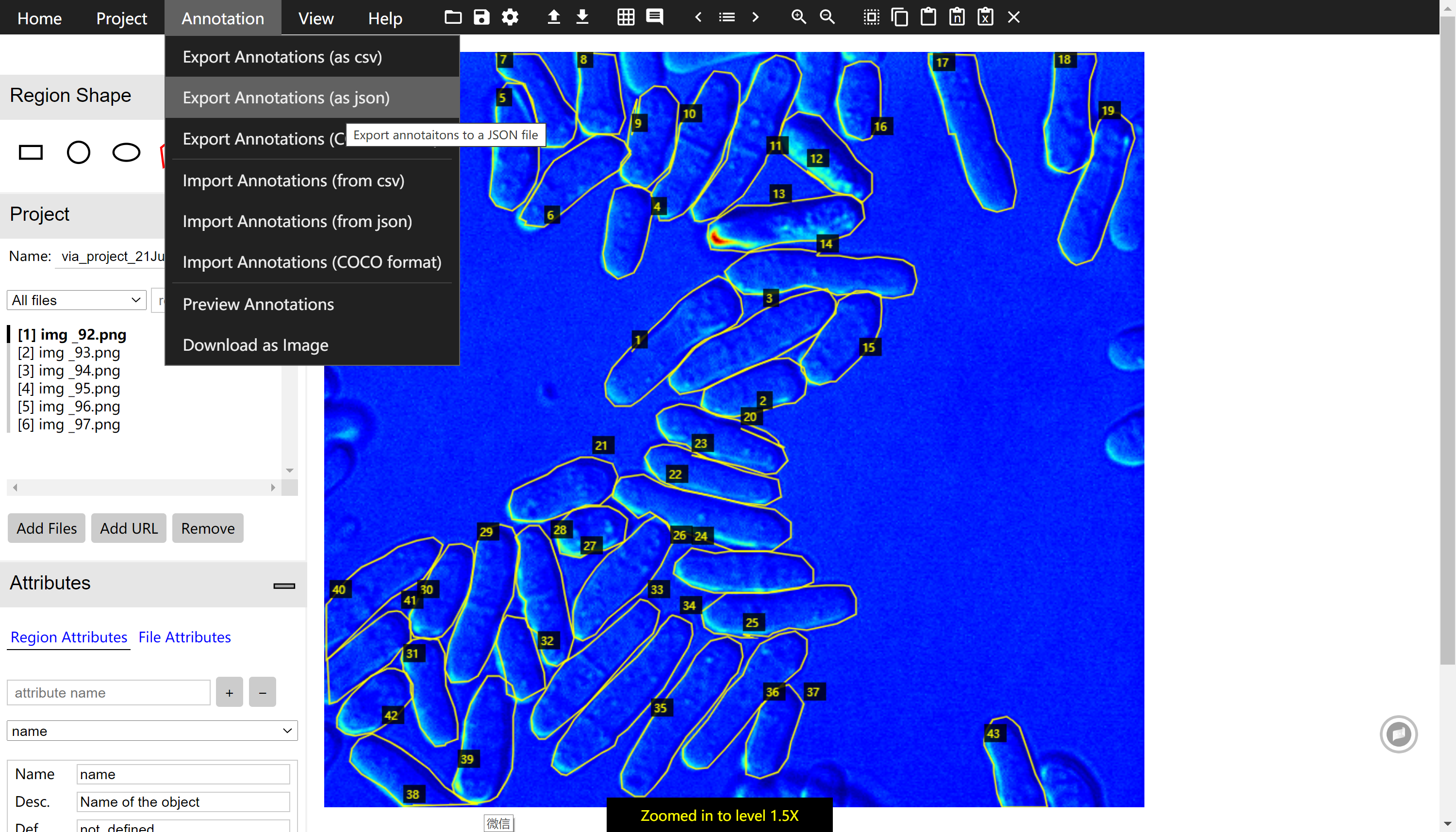
导出文件
标注完成之后需要将标注好的文件进行保存,这里将文件保存为json格式,然后与图片集放置在一起,如果分组进行标记工作,那么每个人都会导出一个json文件,只要最后将json文件和图片集汇总然后重新导入VIA生成即可。
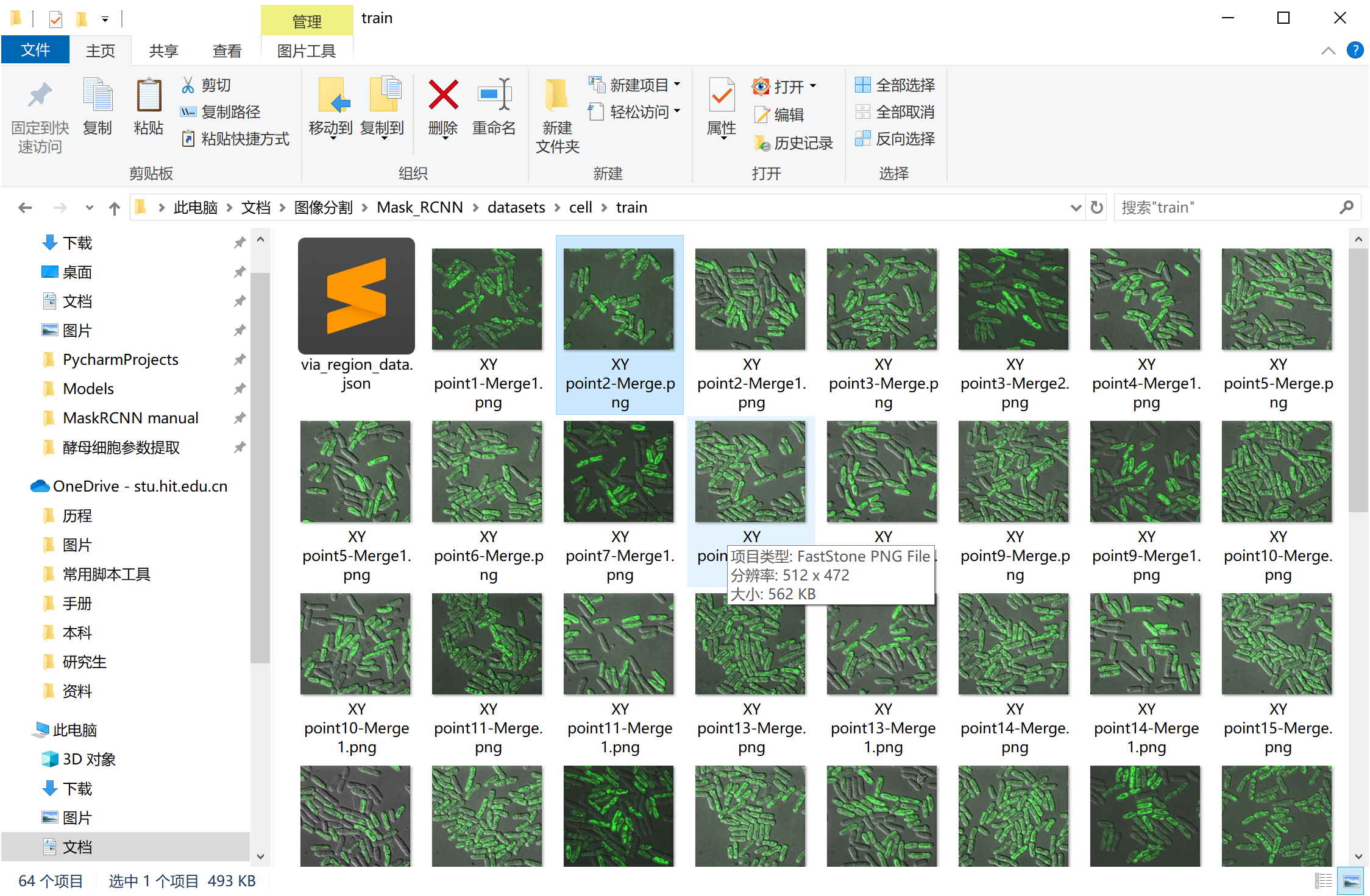
存储数据集
在Mask R-CNN项目的datasets中新建cell 目录, 然后将VIA汇总得到的json和图片放置在该目录下。其中train和val分目录存储。
代码编写
在Mask_RCNN/samples下新建cell目录,仿照nucleus编写主程序。
环境安装
该项目的主要环境依赖如下所示,实际运行时可根据提示进行相应库的安装, 本机(CPU)版本下正常运行的环境配置可见附录。
xxxxxxxxxx# 显卡:NVIDIA 1080Ti# 内存:>8GB# 环境依赖:## cudatoolkit==10.0.130## cudnn==7.6.5## tensorflow=1.14.0## keras==2.2.4## pillow==5.3.0## opencv-python==3.4.2## scikit-image==0.16.2## numpy==1.18.4## imgaug## PyQt5## matplotlib==3.1.1模型训练
采用迁移学习的方法,基于COCO数据集(千万张图片)训练得到的权重,保持convolutional backbone部分权重不变,导入细胞数据集,调整Head部分的权重,训练60代,得到用于识别酵母细胞的模型。下载[预训练的权重文件][https://github.com/lizhogn/Cell_Analysis/releases].
模型预测
输入测试图像,模型预测输出如下信息:
xxxxxxxxxxpredict_out:{ class_id: [num_cells,], # 代表识别对象的类别号,当前只有一类细胞,故类别号均为1.(未来可以识别多类细胞) scrores: [num_cells,], # 代表识别对象的准确度,有多大概率保证。 rois: [num_cells, 4], # 代表识别对象的区域值,由(x1,y1,x2,y2)对角线坐标确定 masks: [height, width, num_cells] # 代表识别对象的掩膜图像,和原始图像的长宽一致}- 通过掩膜可进行细胞的分离:

通过rois和掩膜可进行检测结果的绘制
附录
本机环境配置文件:
xxxxxxxxxx# 生成# conda env export > environment.yaml # 重建# conda env create -f environment.yamlnamemaskrcnnchannelsdefaultsdependencies_libgcc_mutex=0.1=main_tflow_select=2.3.0=mklabsl-py=0.9.0=py36_0astor=0.8.1=py36_0blas=1.0=mklc-ares=1.15.0=h7b6447c_1001ca-certificates=2020.6.24=0certifi=2020.6.20=py36_0dbus=1.13.16=hb2f20db_0expat=2.2.9=he6710b0_2fontconfig=2.13.0=h9420a91_0freetype=2.10.2=h5ab3b9f_0gast=0.3.3=py_0glib=2.65.0=h3eb4bd4_0grpcio=1.31.0=py36hf8bcb03_0gst-plugins-base=1.14.0=hbbd80ab_1gstreamer=1.14.0=hb31296c_0h5py=2.10.0=py36hd6299e0_1hdf5=1.10.6=hb1b8bf9_0icu=58.2=he6710b0_3importlib-metadata=1.7.0=py36_0intel-openmp=2020.1=217jpeg=9b=h024ee3a_2keras-applications=1.0.8=py_1keras-preprocessing=1.1.0=py_1ld_impl_linux-64=2.33.1=h53a641e_7libedit=3.1.20191231=h14c3975_1libffi=3.3=he6710b0_2libgcc-ng=9.1.0=hdf63c60_0libgfortran-ng=7.3.0=hdf63c60_0libpng=1.6.37=hbc83047_0libprotobuf=3.6.0=hdbcaa40_0libstdcxx-ng=9.1.0=hdf63c60_0libuuid=1.0.3=h1bed415_2libxcb=1.14=h7b6447c_0libxml2=2.9.10=he19cac6_1markdown=3.2.2=py36_0mkl=2020.1=217mkl-service=2.3.0=py36he904b0f_0mkl_fft=1.1.0=py36h23d657b_0mkl_random=1.1.1=py36h0573a6f_0ncurses=6.2=he6710b0_1numpy=1.19.1=py36hbc911f0_0numpy-base=1.19.1=py36hfa32c7d_0openssl=1.1.1g=h7b6447c_0pcre=8.44=he6710b0_0pip=20.2.2=py36_0protobuf=3.6.0=py36hf484d3e_0pyqt=5.9.2=py36h05f1152_2python=3.6.10=h7579374_2qt=5.9.7=h5867ecd_1readline=8.0=h7b6447c_0scipy=1.5.0=py36h0b6359f_0setuptools=49.6.0=py36_0sip=4.19.8=py36hf484d3e_0six=1.15.0=py_0sqlite=3.32.3=h62c20be_0tensorboard=1.11.0=py36hf484d3e_0tensorflow=1.11.0=mkl_py36ha6f0bda_0tensorflow-base=1.11.0=mkl_py36h3c3e929_0termcolor=1.1.0=py36_1tk=8.6.10=hbc83047_0werkzeug=1.0.1=py_0wheel=0.34.2=py36_0xz=5.2.5=h7b6447c_0zipp=3.1.0=py_0zlib=1.2.11=h7b6447c_3pipaltgraph==0.17backcall==0.2.0cycler==0.10.0decorator==4.4.2imageio==2.9.0imutils==0.5.3ipython==7.16.1ipython-genutils==0.2.0jedi==0.17.2keras==2.2.4kiwisolver==1.2.0matplotlib==3.1.1networkx==2.4opencv-python==4.4.0.42parso==0.7.1pexpect==4.8.0pickleshare==0.7.5pillow==7.2.0prompt-toolkit==3.0.6ptyprocess==0.6.0pygments==2.6.1pyinstaller==4.0pyinstaller-hooks-contrib==2020.7pyparsing==2.4.7pyqt5-sip==12.8.0pyqtgraph==0.11.0python-dateutil==2.8.1pywavelets==1.1.1pyyaml==5.3.1scikit-image==0.17.2tifffile==2020.8.13traitlets==4.3.3wcwidth==0.2.5prefix/home/lizhogn/anaconda3/envs/maskrcnnxxxxxxxxxx# pip 安装# 导出:pip freeze > requirement.txt# 恢复:pip install -r requirement.txt -i https://pypi.tuna.tsinghua.edu.cn/simple# requirement.txtabsl-py==0.9.0altgraph==0.17astor==0.8.1backcall==0.2.0certifi==2020.6.20cycler==0.10.0decorator==4.4.2gast==0.3.3h5py==2.10.0imageio==2.9.0imutils==0.5.3ipython==7.16.1ipython-genutils==0.2.0jedi==0.17.2Keras==2.2.4kiwisolver==1.2.0matplotlib==3.1.1networkx==2.4numpy==1.19opencv-python==4.4.0.42parso==0.7.1pexpect==4.8.0pickleshare==0.7.5Pillow==7.2.0prompt-toolkit==3.0.6protobuf==3.6.0ptyprocess==0.6.0Pygments==2.6.1pyinstaller==4.0pyparsing==2.4.7pyqt5PyQt5-sip==12.8.0pyqtgraph==0.11.0python-dateutil==2.8.1PyWavelets==1.1.1PyYAML==5.3.1scikit-image==0.15.1scipy==1.5.0six==1.15.0tensorboard==1.11.0tensorflow==1.11.0termcolor==1.1.0tifffile==2020.8.13traitlets==4.3.3wcwidth==0.2.5Werkzeug==1.0.1zipp==3.1.0
细胞参数提取
细胞参数分两方面进行提取:
- 细胞形态参数:长度、宽度
- 线粒体参数:线粒体总长度、段数、各段长度
细胞形态参数
利用Mask R-CNN模型输出结果中的掩膜图像,可进行细胞形态参数(长度、宽度)的提取。以一个细胞的提取进行演示:
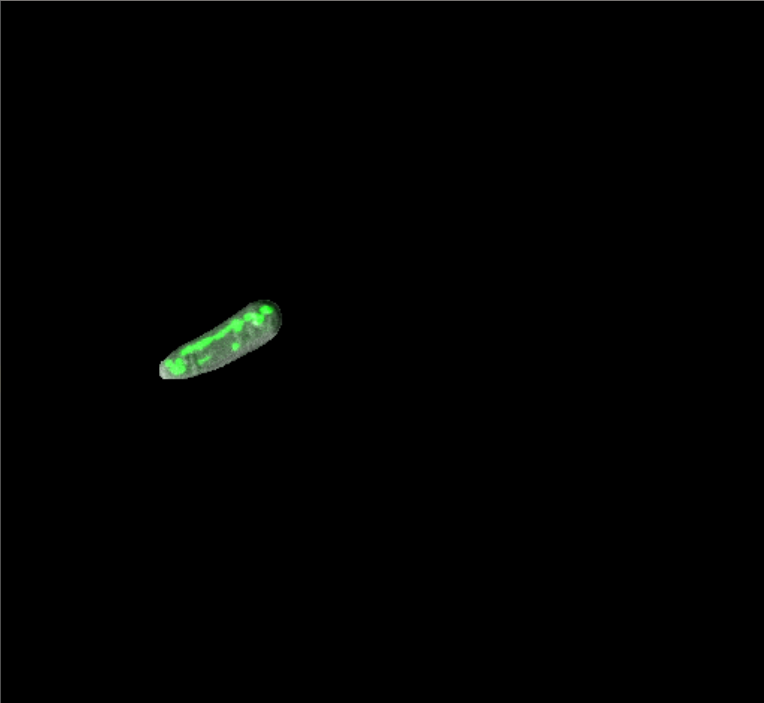 |
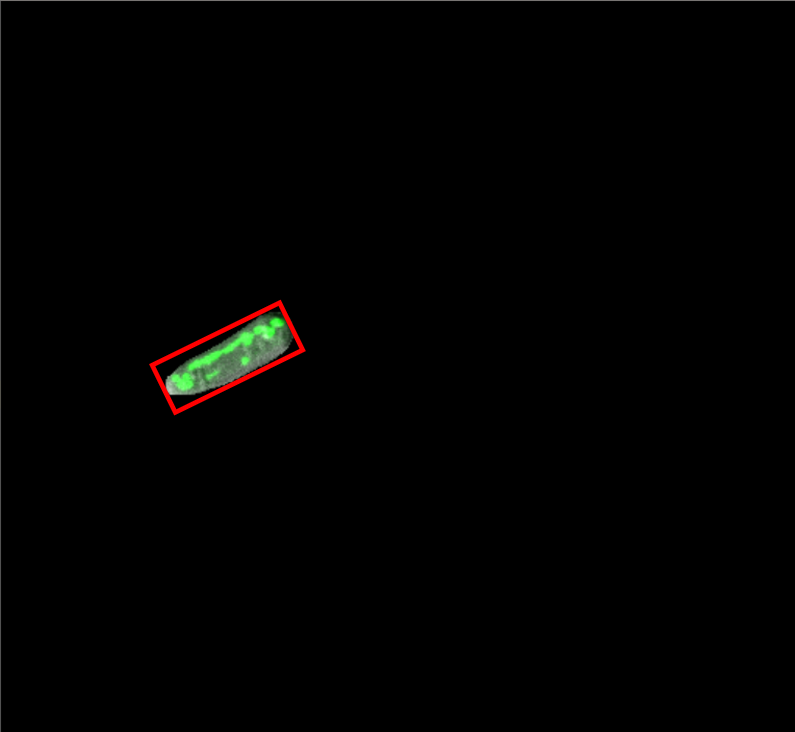 |
 |
| 图1.细胞掩膜 | 图2.正外接矩形 | 图3.最小外接矩形 |
图1便是由Mask R-CNN识别的单个细胞,在掩膜数组中数值只有 0 和 1(非0数即为1)。酵母细胞的形状近似于一个狭长的椭球状,长宽差异明显,并且沿中轴线无明显的弯曲,所以可以用一个最小的外接细胞矩形的长和宽来代替细胞的长和宽,如图3所示,这个外接矩形的学术名叫做连通域最小外接矩形。求该矩形坐标及旋转角度的算法如下:
第一步:正外接矩形的算法
xxxxxxxxxx# 伪代码# 算法原理:检测非0像素的位置坐标的最小点和最大点Def Find_Bounding_Box(mask_img): 初始化X_MIN, X_MAX, Y_MIN, Y_MAX FOR X,Y IN 掩膜中所有为‘1’的坐标点 IF X < X_MIN: X_MIN = X ELIF X > X_MAX: X_MAX = X ELIF Y < Y_MIN: Y_MIN = Y ELIF Y > Y_MAX: Y_MAX = Y # 正外接矩形的左上角坐标点: LEFT_TOP = (X_MIN, Y_MIN) # 正外接矩形右下角坐标点: RIGHT_BOTTOM = (X_MAX, Y_MAX) return LEFT_TOP, RIGHT_BOTTOM第二步:最小外接矩形的确定
xxxxxxxxxx# 伪代码# 算法原理:将图像进行旋转,每旋转1°, 检测一次正外接矩形,计算非0区域面积和矩形区域面积的比值,取出比值最大的正外接矩形和旋转角度Def Find_MinArea_Box(mask_img): mask_area = sum(mask_img) 初始化area_min, point1, point2, angle FOR ANGLE IN RANGE(0, 180): IMG = ROTATE(mask_img, ANGLE) POINT1, POINT2 = Find_Bounding_Box(IMG) box_area = (POINT2.X - POINT1.X ) * (POINT2.Y - POINT2.Y) IF AREA_MIN > mask_area / box_area angle = ANGLE point1 = POINT1 point2 = POINT2 # 端点旋转换算 point1, point2 = DEROTATE(point1, point2, -angle) return point1, point2, angle在Opencv-python中,最小外接矩形算法由minAreaRect进行封装,可以直接调用。
最终可以输出最小外接矩形的坐标,进而求出细胞的长度和宽度。

线粒体形态参数

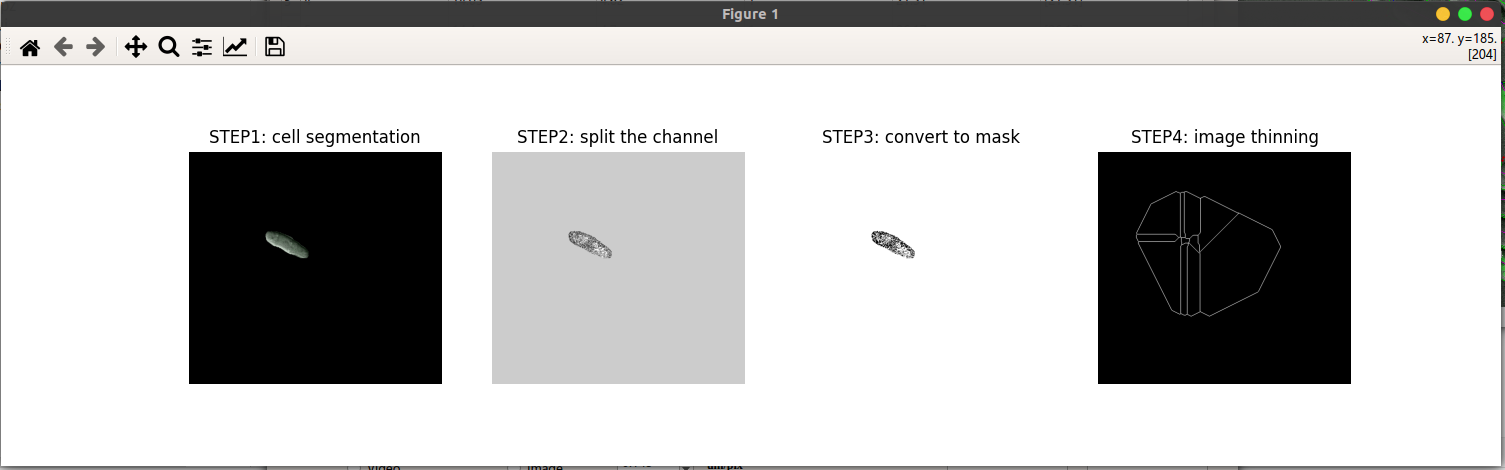
线粒体参数(线粒体数目、各段长度、总长度)提取流程如上图所示,
第一步,从细胞实例分割的结果中分理出单个细胞;
xxxxxxxxxxsingleCell = img * mask[i]第二步,将图中的绿色通道分离出来,并进行对比度的增强。
xxxxxxxxxx# 原理:观察下图可以发现在非细胞区域同样存在着一定量的绿色分量,因此不能简单的保留绿色通道。但可以发现非细胞区域三个通道分量的值差别不大。# 算法:red_channel = img[:,:,0]green_channel = img[:,:,1]blue_channel = img[:,:,2]mitochondrial_img = green_channel - (red_channel+blue_channel)/2contrast_img = adjustBrightness(mitochondrial_img)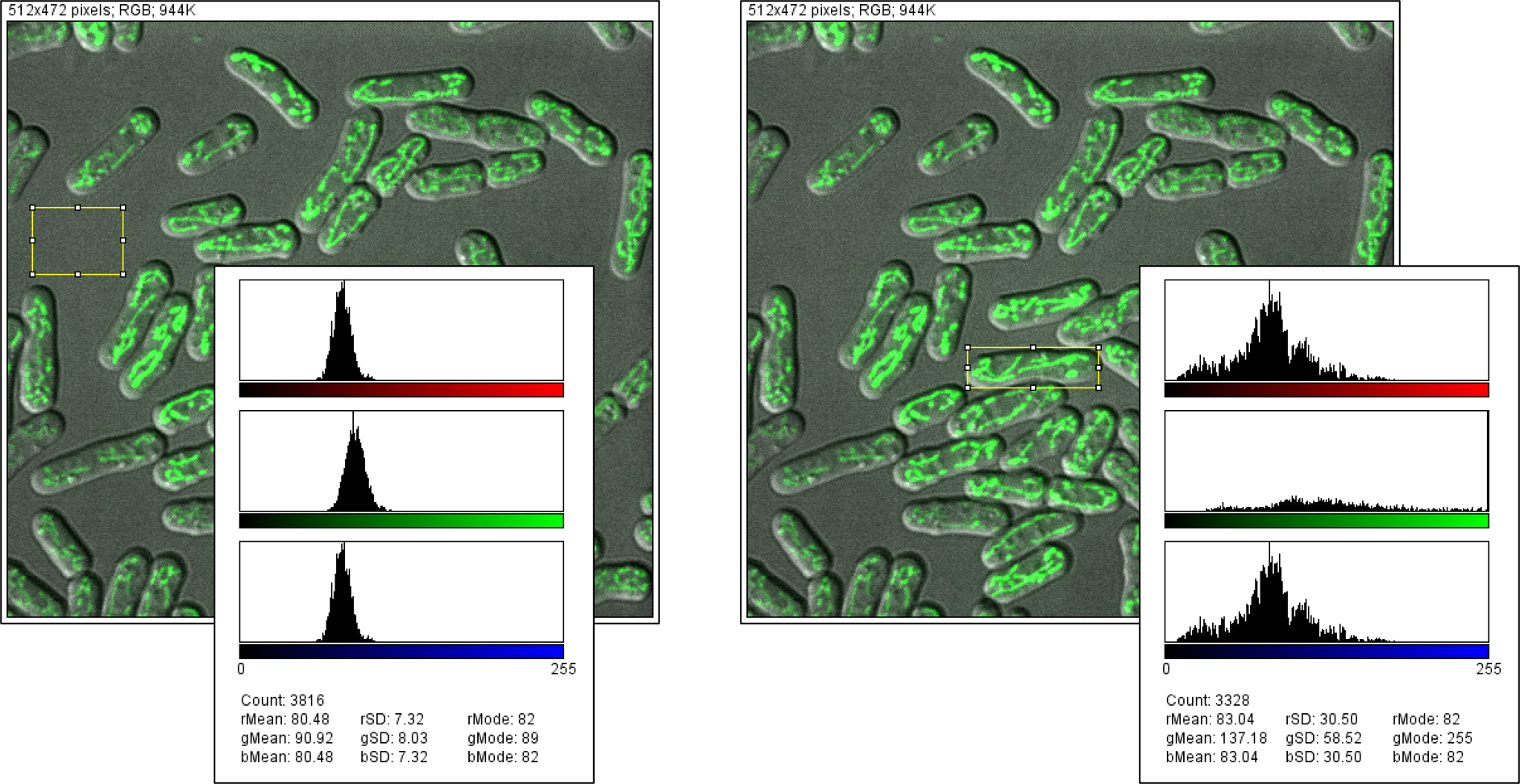
- 第三步,进行图像的阈值分割
原理:OTSU算法:

- 将灰度值区间为[0,m],对于[0,m]间的每一个灰度t,将它作为阈值将图像分割为灰度为[0,t]以及[t+1,m]两部分。
- 计算每一部分的所占比例,每一部分的平均灰度值,以及总的平均灰度值。
- 计算他们的类间方差
- 取出类间方差最大时对应的阈值t,这就可以作为我们最终所取的阈值。
第四步,进行图像的细化操作
细化是一种形态学操作,用于从二进制图像中去除选定的前景像素,有点像侵蚀或开放。它可以用于多个应用程序,但对于骨架化特别有用。在这种模式下,通常采用将边缘检测器的输出减小到单像素厚度的方法进行整理。细化通常只适用于二值图像,并产生另一个二值图像作为输出。
关于图像细化的原理不做展开分析,相关原理可以参见冈萨雷斯和伍兹: 《数字图像处理》、和这篇[博文][https://homepages.inf.ed.ac.uk/rbf/HIPR2/thin.htm]。在
scikit-image的morphology模块中,可以直接进行图像细化算法的调用
线粒体个数-总长度-各段长度
线粒体个数:等于细化二值图像连通域的个数,相关函数:cv2.measure.label
总长度:等于细化二值图像的面积,求和即可。
各段长度:等于细化二值图像各个连通域的面积,相关函数:cv2.measure.regionprops
线粒体分支节点检测
任意一张线粒体骨架图片,其中的分支节点与8-邻域的位置是固定的,通过统计,可以总结为如下40种情况(其中白色像素代表1, 黑色像素代表0):
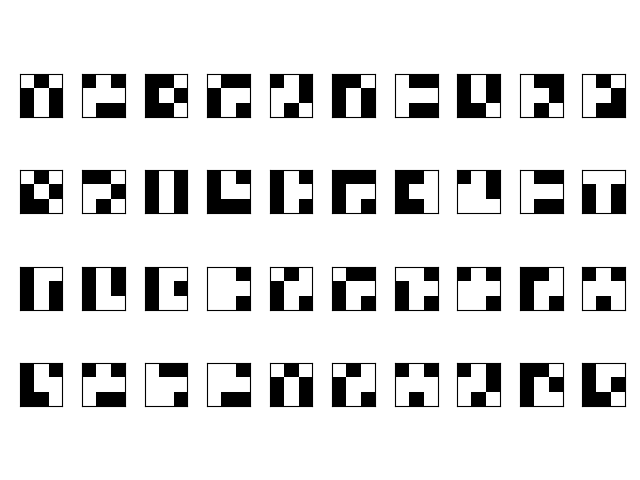
因此,只需要遍历线粒体骨架图片中每一个非零的像素点,然后去匹配这40种情况,最后再去除距离较近的分支点,剩下的便是线粒体分支节点的检测结果。
算法流程如下:
x# 交叉点检测算法# 参考 https://gist.github.com/jamjar919/05a76cf21035cef3cc86ac1979588d6ddef getSkeletonIntersection(skeleton): """ Given a skeletonised image, it will give the coordinates of the intersections of the skeleton. Keyword arguments: skeleton -- the skeletonised image to detect the intersections of Returns: List of 2-tuples (x,y) containing the intersection coordinates """ # A biiiiiig list of valid intersections 2 3 4 # These are in the format shown to the right 1 C 5 # 8 7 6 # 列举交叉节点所有可能的情况 validIntersection = [[0,1,0,1,0,0,1,0],[0,0,1,0,1,0,0,1],[1,0,0,1,0,1,0,0], [0,1,0,0,1,0,1,0],[0,0,1,0,0,1,0,1],[1,0,0,1,0,0,1,0], [0,1,0,0,1,0,0,1],[1,0,1,0,0,1,0,0],[0,1,0,0,0,1,0,1], [0,1,0,1,0,0,0,1],[0,1,0,1,0,1,0,0],[0,0,0,1,0,1,0,1], [1,0,1,0,0,0,1,0],[1,0,1,0,1,0,0,0],[0,0,1,0,1,0,1,0], [1,0,0,0,1,0,1,0],[1,0,0,1,1,1,0,0],[0,0,1,0,0,1,1,1], [1,1,0,0,1,0,0,1],[0,1,1,1,0,0,1,0],[1,0,1,1,0,0,1,0], [1,0,1,0,0,1,1,0],[1,0,1,1,0,1,1,0],[0,1,1,0,1,0,1,1], [1,1,0,1,1,0,1,0],[1,1,0,0,1,0,1,0],[0,1,1,0,1,0,1,0], [0,0,1,0,1,0,1,1],[1,0,0,1,1,0,1,0],[1,0,1,0,1,1,0,1], [1,0,1,0,1,1,0,0],[1,0,1,0,1,0,0,1],[0,1,0,0,1,0,1,1], [0,1,1,0,1,0,0,1],[1,1,0,1,0,0,1,0],[0,1,0,1,1,0,1,0], [0,0,1,0,1,1,0,1],[1,0,1,0,0,1,0,1],[1,0,0,1,0,1,1,0], [1,0,1,1,0,1,0,0]]; image = skeleton.copy(); image = image/255; intersections = list(); # 遍历每一个非零像素点 for x in range(1,len(image)-1): for y in range(1,len(image[x])-1): # If we have a white pixel if image[x][y] == 1: # 获取该像素点的8邻域像素值 neighbours = neighbours(x,y,image); valid = True; # 与有效交叉点进行比对 if neighbours in validIntersection: intersections.append((y,x)); # Filter intersections to make sure we don't count them twice or ones that are very close together # 如果检测到的交叉点之间距离小于10个像素,则删除其中的一个交叉点 for point1 in intersections: for point2 in intersections: if (((point1[0] - point2[0])**2 + (point1[1] - point2[1])**2) < 10**2) and (point1 != point2): intersections.remove(point2); # Remove duplicates intersections = list(set(intersections)); # 返回交叉点的坐标 return intersections;相关问题及解决方案
漏检测细胞
细胞漏检测的原因主要有两个流程:
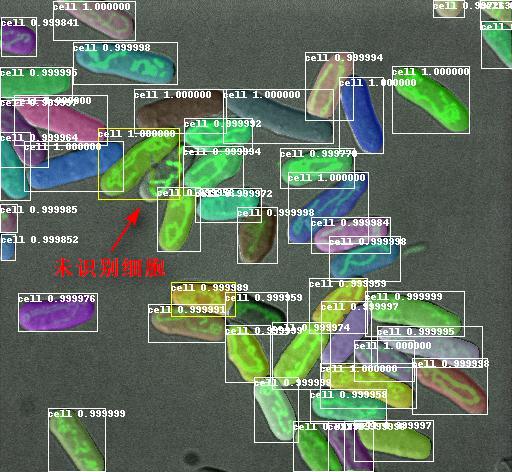
① 首先是Mask R-CNN的网络输出,由于网络训练不充分、非极大值抑制等等的因素,导致有些细胞并不能被检测到。下图中红色箭头所指的位置便是Mask R-CNN网络未检测到的细胞,主要是由于该细胞与左侧的细胞挨得太近导致两个细胞边界框的交并比(IoU)太大而被认同为一个目标,非极大值抑制算法移除了概率较小的那个细胞。
xxxxxxxxxx解决方案:提高非极大值抑制阈值,加大训练样本数目
② 其次是最小矩形框算法略过的矩形框。最小矩形框算法在针对一些细胞的掩膜时会失效,算法报错从而导致程序的终止,
xxxxxxxxxx解决方案:为了能够让程序继续处理后续的细胞,这里采取的策略是按照原来的正外接矩形框进行画框。
长宽测量误差
① 在长度和宽度的测量中,依据的原理是细胞掩膜图像的最小矩形框。而利用Mask R-CNN分割细胞得到的掩膜图像有时候并不是那么理想(如下图所示),会导致细胞的宽度测量出现误差。另外,有些细胞生成的掩膜图像无法应用最小矩形框的算法,当前设定的程序是跳过该细胞长宽的检测,在后续需要完善。
xxxxxxxxxx解决方案:待解决

② 在图像边缘的区域,细胞的长宽测量必然是不准确的。
xxxxxxxxxx解决方案:过滤边界细胞算法:Mask R-CNN输出的roi中任意一条矩形框位于图像边界(1个像素)时,跳过此细胞形态参数的分析。
线粒体测量问题
① 未染色线粒体测量误差,如下图所示,由于某些细胞的线粒体并没有被染色,所以当进行绿色通道分离、阈值分割后,并不能很好的提取线粒体的形态掩膜(本来就没有)。当进行图像细化算法的分析时,所细化的线条也不符合预期。在后续的程序中应该能够有效的识别这些未被染色的细胞,跳过对他们的线粒体形态参数统计过程。
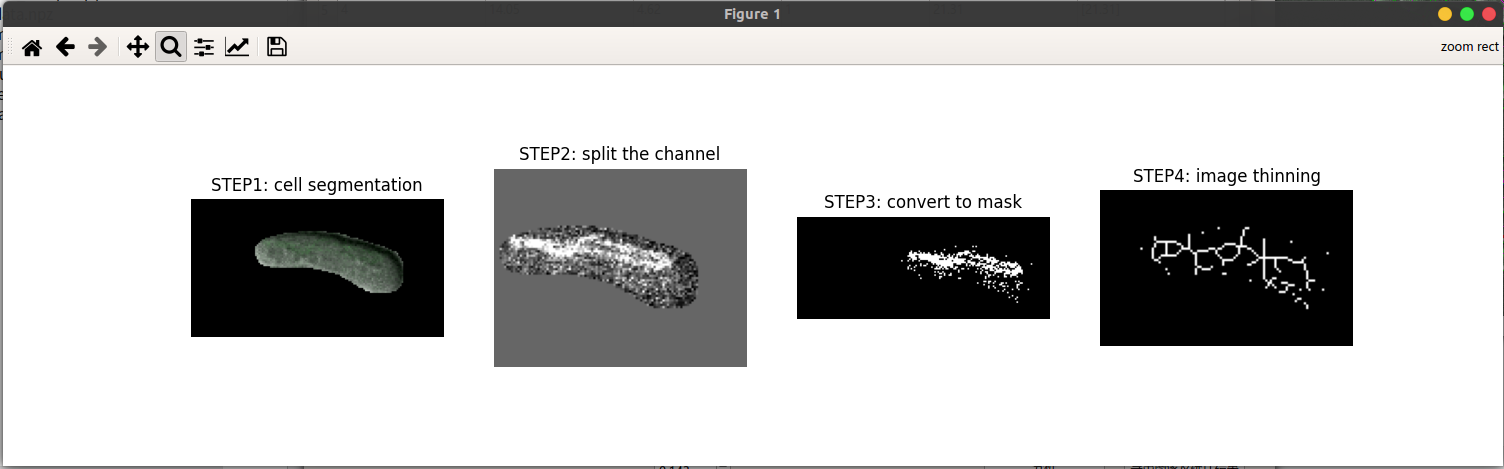
xxxxxxxxxx# 解决方案:跳过此类细胞的线粒体参数提取# 算法:根据绿色分量和其他两者分量之间的差异性,若差异性小,说明该细胞线粒体未被染色。# 放上源代码def _is_miss_mitochondrial(self, img, mask): # judge whether there are mitochondrial in the cell mask_int = mask.astype('uint8') mask_int[mask_int==1] = 255 rhist = cv2.calcHist([img], [0], mask_int, [256], [0, 256]) ghist = cv2.calcHist([img], [1], mask_int, [256], [0, 256]) bhist = cv2.calcHist([img], [2], mask_int, [256], [0, 256]) def weight_average(hist): weights = list(range(256)) mean = sum([hist[i]*i for i in range(256)])/sum(hist) return mean rhist_mean = weight_average(rhist) ghist_mean = weight_average(ghist) bhist_mean = weight_average(bhist) if ghist_mean - (rhist_mean + bhist_mean) / 2 > 20: # Mitochondria in the cell are not lost return False else: # Mitochondria in the cell are lsot return True② 阈值分割以及图像细化过程产生的误差。观察下列线粒体骨架的生成过程,可以发现,由于存在中间的不连续点,程序将它分成了两条线粒体,加上左边小的不连续点,总共得到了四条线粒体。通过过滤,保留了图中长的两条线粒体,像素的个数 × 单位像素的长度 就是线粒体的长度。
xxxxxxxxxx解决方案:系统误差,无法避免。
图形界面开发
该项目的图形界面是基于PyQt5,结合QtDesigner界面布局软件和pyqtgraph画图模块进行开发的,通过API调用实现图形与算法的分离。
实现:
文件导入、图片预览、结果导出
细胞处理分析
批量处理
尚未实现:
多线程
xxxxxxxxxx# 程序主入口:main.py# -*- coding: utf-8 -*-import sysimport osimport csvimport warnings# ignore the warningswarnings.filterwarnings('ignore')import cv2from PyQt5 import uic, QtWidgetsfrom PyQt5.QtWidgets import QFileDialog, QMainWindow, QHeaderViewfrom PyQt5.QtWidgets import QMessageBox, QAbstractItemView, QTableWidgetItemfrom PyQt5 import QtCorefrom PyQt5.QtCore import QThread, pyqtSignalimport pyqtgraph as pgpg.setConfigOption('imageAxisOrder', 'row-major')from cell_analysis import InferenceConfigfrom cell_analysis import cell_analysisfrom mrcnn import model as modellibQtCore.QCoreApplication.setAttribute(QtCore.Qt.AA_EnableHighDpiScaling)class PyQtMainEntry(QMainWindow): def __init__(self): super().__init__() # load the ui from the file self.ui = uic.loadUi("ui/main.ui") self.result_window = uic.loadUi("ui/result.ui") # initialize the signal/slot self.signal_init() # initialize the model self.model_init() def signal_init(self): # initialize the signal/slot function # mainWindow signal initialized self.ui.browser_btn.clicked.connect(self.file_Browser_Clicked) self.ui.analysis_btn.clicked.connect(self.startAnalysis_Clicked) self.ui.export_btn.clicked.connect(self.export_Clicked) self.ui.add_btn.clicked['bool'].connect(self.add_item_Clicked) self.ui.sub_btn.clicked.connect(self.sub_item_Clicked) self.ui.clear_btn.clicked.connect(self.clear_item_Clicked) self.ui.filelist.itemClicked['QListWidgetItem*'].connect(self.item_Clicked) # result window signal initialized self.result_window.check_btn.clicked.connect(self.check_Clicked) self.result_window.export_table_btn.clicked.connect(self.export_table_Clicked) def model_init(self): # load the maskrcnn model config = InferenceConfig() self.model = modellib.MaskRCNN(mode='inference', config=config, model_dir='logs') weights_path = 'logs/cell/mask_rcnn_cell_0060.h5' print("Loading weights ", weights_path) self.model.load_weights(weights_path, by_name=True) print('Load the model sucessfully') def file_Browser_Clicked(self): print('file_Browser_Clicked clicked') self.files, _ = QFileDialog.getOpenFileNames(self, '打开图片') self.ui.lineEdit.setText(os.path.dirname(self.files[0])) files_basename = [os.path.basename(x) for x in self.files] self.ui.filelist.addItems(files_basename) # show the first image in the ImageViewer self.img = cv2.imread(self.files[0]) self.ui.graphWidget.setImage(self.img) def startAnalysis_Clicked(self): # start analysis the select img self.result_window.show() print('分析中...') # *********************************************************************************************** self.cell_obj = cell_analysis(self.model, self.img, calibration=self.ui.doubleSpinBox.text()) # show img self.result_window.widget.setImage(self.cell_obj.detect_img) # show cell count result table self.set_result_table(self.cell_obj.merge_measurements()) # ************************************************************************************************** # start a thread to analysis # self.analysis_thread = WorkThread(self.img, self.model, self.ui.doubleSpinBox.text()) # self.analysis_thread.start() # self.analysis_thread.trigger.connect(self.show_results) # ************************************************************************************************** def show_results(self, cell_obj): # show img self.result_window.widget.setImage(cell_obj.detect_img) # show cell count result table self.set_result_table(cell_obj.merge_measurements()) def set_result_table(self, data): # set the result table in the result window widget cow_label = list(data[0].keys()) cow_name = ['细胞ID', '长度(um)','宽度(um)', '线粒体个数', '总长度(um)', '各段长度(um)'] cow_number = len(data[0]) row_number = len(data) self.result_window.tableWidget.setColumnCount(cow_number) self.result_window.tableWidget.setRowCount(row_number) # se the header label in the horizontal direction and the header label in the vertical direction self.result_window.tableWidget.setHorizontalHeaderLabels(cow_name) # set the horizontal diretion table to the adaptive stretch mode self.result_window.tableWidget.horizontalHeader().setSectionResizeMode(QHeaderView.Stretch) # set the entire row of the table to be selected self.result_window.tableWidget.setSelectionBehavior(QAbstractItemView.SelectRows) # set full of the widget self.result_window.tableWidget.horizontalHeader().setStretchLastSection(True) # add data into the table for row in range(row_number): for cow in range(cow_number): cur_cell = data[row] print(cur_cell[cow_label[cow]]) newItem = QTableWidgetItem(str(cur_cell[cow_label[cow]])) self.result_window.tableWidget.setItem(row, cow, newItem) def export_Clicked(self): print('export_Clicked clicked') output_dir = QFileDialog.getExistingDirectory(self, 'Choose a Directory to save result') if output_dir: # batch process print(self.files) for file in self.files: img = cv2.imread(file) cell_obj = cell_analysis(self.model, img, calibration=self.ui.doubleSpinBox.text()) # 1. mkdir dir_path = os.path.join(output_dir, os.path.basename(file).split('.')[0]) print('current path dir', dir_path) if not os.path.exists(dir_path): os.mkdir(dir_path) # 1. origin img save origin_img_name = os.path.join(dir_path, 'origin.png') cv2.imwrite(origin_img_name, img=img) # 2. detect img save detect_img_name = os.path.join(dir_path, 'detect.png') cv2.imwrite(detect_img_name, img=cell_obj.detect_img) # 3. cell morphological and mitochondrial infos save cell_infos = cell_obj.merge_measurements() cell_infos_name = os.path.join(dir_path, 'cell_infos.csv') with open(cell_infos_name, 'w', newline='') as csvfile: fieldnames = list(cell_infos[0].keys()) fieldnames = ['Cell_Id', 'Length(um)', 'Width(um)', 'mitochondrial_numbers', 'mitochondrial_overall_length(um)', 'mitochondrial_each_segment_len(um)'] writer = csv.DictWriter(csvfile, fieldnames=fieldnames) writer.writeheader() writer.writerows(cell_infos) print('save sucessfully') QMessageBox.information(self, '提示', 'Save sucessfully!') # self.setCentralWidget(QMessageBox) def add_item_Clicked(self): # while the add button "+" pressed # open the file dialog and append new img into filelistWidget files, _ = QFileDialog.getOpenFileNames(self, '选择图片') self.ui.lineEdit.setText(os.path.dirname(files[0])) file_basename = [os.path.basename(x) for x in files] self.ui.filelist.addItems(file_basename) def sub_item_Clicked(self): # while the add button "-" pressed # delete current selected items for item in self.ui.filelist.selectedItems(): self.ui.filelist.takeItem(self.ui.filelist.row(item)) def clear_item_Clicked(self): print('clear_item_Clicked clicked') # while "clear" button is cliked, clear the list self.ui.filelist.clear() def item_Clicked(self): # if item clicked, change the ImageView content to current select image print('item_Clicked clicked') cur_filename = self.ui.filelist.selectedItems()[0].text() print(cur_filename) cur_filename = os.path.join(self.ui.lineEdit.text(), cur_filename) self.img = cv2.imread(cur_filename) self.ui.graphWidget.setImage(self.img) def check_Clicked(self): # print('check_Clicked clicked') # plot the current row responding cell process cur_cell_id = self.result_window.tableWidget.currentRow() self.cell_obj.show_specific_cell(cell_id=cur_cell_id) def export_table_Clicked(self): # print('export table clicked') # export the result table and img output_dir = QFileDialog.getExistingDirectory(self, 'Open Directory to save the result') # result save if output_dir: try: # 1. origin img save origin_img_name = os.path.join(output_dir, 'origin.png') cv2.imwrite(origin_img_name, img=self.img) # 2. detect img save detect_img_name = os.path.join(output_dir, 'detect.png') cv2.imwrite(detect_img_name, img=self.cell_obj.detect_img) # 3. cell morphological and mitochondrial infos save cell_infos = self.cell_obj.merge_measurements() cell_infos_name = os.path.join(output_dir, 'cell_infos.csv') with open(cell_infos_name, 'w', newline='') as csvfile: fieldnames = list(cell_infos[0].keys()) fieldnames = ['Cell_Id', 'Length(um)', 'Width(um)', 'mitochondrial_numbers', 'mitochondrial_overall_length(um)', 'mitochondrial_each_segment_len(um)'] writer = csv.DictWriter(csvfile, fieldnames=fieldnames) writer.writeheader() writer.writerows(cell_infos) print('save sucessfully') QMessageBox.information(self, '提示', 'Save sucessfully!') except: QMessageBox.information(self, '提示', 'Save Failure, Please check your save directory!')class WorkThread(QThread): trigger = pyqtSignal(object) def __init__(self, img, model, calibration): super(WorkThread, self).__init__() self.img = img self.model = model self.calibration = calibration def run(self): print(self.img) print(self.model) print(self.calibration) self.cell_obj = cell_analysis(self.model, self.img, calibration=self.calibration) self.trigger.emit(self.cell_obj)if __name__ == "__main__": app = QtWidgets.QApplication(sys.argv) window = PyQtMainEntry() window.ui.show() sys.exit(app.exec_())软件发布
为了使软件脱离python特定环境而在计算机上独立运行,需要将py程序打包为二进制可执行文件,现在比较常用的打包工具为PyInstaller。
安装
首先需要安装PyInstaller, 安装方式如下:
xxxxxxxxxx$ pip install pyinstaller -i https://pypi.tuna.tsinghua.edu.cn/simple打包
PyInstaller打包使用语法如下:
xxxxxxxxxx(maskrcnn) ➜ ~ pyinstaller -husage: pyinstaller [-h] [-v] [-D] [-F] [--specpath DIR] [-n NAME] [--add-data <SRC;DEST or SRC:DEST>] [--add-binary <SRC;DEST or SRC:DEST>] [-p DIR] [--hidden-import MODULENAME] [--additional-hooks-dir HOOKSPATH] [--runtime-hook RUNTIME_HOOKS] [--exclude-module EXCLUDES] [--key KEY] [-d {all,imports,bootloader,noarchive}] [-s] [--noupx] [--upx-exclude FILE] [-c] [-w] [-i <FILE.ico or FILE.exe,ID or FILE.icns>] [--version-file FILE] [-m <FILE or XML>] [-r RESOURCE] [--uac-admin] [--uac-uiaccess] [--win-private-assemblies] [--win-no-prefer-redirects] [--osx-bundle-identifier BUNDLE_IDENTIFIER] [--runtime-tmpdir PATH] [--bootloader-ignore-signals] [--distpath DIR] [--workpath WORKPATH] [-y] [--upx-dir UPX_DIR] [-a] [--clean] [--log-level LEVEL] scriptname [scriptname ...]各参数意义如下:
| 参数 | 说明 |
|---|---|
| -F, –onefile | 打包一个单个文件,如果你的代码都写在一个.py文件的话,可以用这个,如果是多个.py文件就别用 |
| -D, –onedir | 打包多个文件,在dist中生成很多依赖文件,适合以框架形式编写工具代码,我个人比较推荐这样,代码易于维护 |
| -K, –tk | 在部署时包含 TCL/TK |
| -a, –ascii | 不包含编码.在支持Unicode的python版本上默认包含所有的编码. |
| -d, –debug | 产生debug版本的可执行文件 |
| -w,–windowed,–noconsole | 使用Windows子系统执行.当程序启动的时候不会打开命令行(只对Windows有效) |
| -c,–nowindowed,–console | 使用控制台子系统执行(默认)(只对Windows有效)pyinstaller -c xxxx.pypyinstaller xxxx.py --console |
| -s,–strip | 可执行文件和共享库将run through strip.注意Cygwin的strip往往使普通的win32 Dll无法使用. |
| -X, –upx | 如果有UPX安装(执行Configure.py时检测),会压缩执行文件(Windows系统中的DLL也会)(参见note) |
| -o DIR, –out=DIR | 指定spec文件的生成目录,如果没有指定,而且当前目录是PyInstaller的根目录,会自动创建一个用于输出(spec和生成的可执行文件)的目录.如果没有指定,而当前目录不是PyInstaller的根目录,则会输出到当前的目录下. |
| -p DIR, –path=DIR | 设置导入路径(和使用PYTHONPATH效果相似).可以用路径分割符(Windows使用分号,Linux使用冒号)分割,指定多个目录.也可以使用多个-p参数来设置多个导入路径,让pyinstaller自己去找程序需要的资源 |
| –icon=<FILE.ICO> | 将file.ico添加为可执行文件的资源(只对Windows系统有效),改变程序的图标 pyinstaller -i ico路径 xxxxx.py |
| –icon=<FILE.EXE,N> | 将file.exe的第n个图标添加为可执行文件的资源(只对Windows系统有效) |
| -v FILE, –version=FILE | 将verfile作为可执行文件的版本资源(只对Windows系统有效) |
| -n NAME, –name=NAME | 可选的项目(产生的spec的)名字.如果省略,第一个脚本的主文件名将作为spec的名字 |
其中,scriptname为主程序入口,是必须的参数,在本项目中,代码层级如下,程序的主要入口为main.py。
xxxxxxxxxx(maskrcnn) ➜ cell_analysis tree -L 1 Cell\ AnalysisCell Analysis├── cell_analysis.py├── dist├── input_imgs├── logs├── main.py├── mrcnn├── output├── README.md├── ui└── utils故打包的代码为:
xxxxxxxxxx$ pyinstaller -D main.py运行完成之后会在对应目录下生成dist目录,里面便存放着可运行的二进制文件。
常见问题
生成的二进制文件并不能直接运行,可通过终端查看问题所在。
UI 文件和模型路径找不到
原因:PyInstaller在打包python程序只能将代码本身和包含的模块进行封装,至于.ui和.h5文件需要手动添加。添加方式是按照源代码中相对路径的存放方式,将文件放置到对应的位置。
Matplotlib 版本问题
错误提示如下:
xxxxxxxxxxexec(bytecode, module.dict)File "matplotlib/init.py", line 898, inFile "matplotlib/cbook/init.py", line 480, in _get_data_pathFile "matplotlib/init.py", line 239, in wrapperFile "matplotlib/init.py", line 534, in get_data_pathFile "matplotlib/init.py", line 239, in wrapperNameError: name 'defaultParams' is not defined问题在于matplotlib版本太新,PyInstaller(4.0)无法将其打包。解决方式: 降低matplotlib版本。
xxxxxxxxxx$ pip install matplotlib==3.1.1 -i https://pypi.tuna.tsinghua.edu.cn/simple模块缺失
错误提示如下:
xxxxxxxxxxModuleNotFoundError: No module named 'MODULENAME'重新打包程序,命令如下:
xxxxxxxxxx$ pyinstaller -D main.py --hidden-import='MODULENAME'例如:
xxxxxxxxxx$ pyinstaller -D main.py --hidden-import='skimage.feature._orb_descriptor_positions'
添加主窗口图标
程序窗口图标
我们程序运行的窗口,需要显示自己的图标,这样才更像一个正式的产品。
通过如下代码,我们可以把一个png图片文件作为 程序窗口图标。
xxxxxxxxxxfrom PyQt5.QtGui import QIconapp = QApplication([])# 加载 iconapp.setWindowIcon(QIcon('logo.png'))注意:这些图标png文件,在使用PyInstaller创建可执行程序时,也要拷贝到程序所在目录。否则可执行程序运行后不会显示图标。
应用程序图标
应用程序图标是放在可执行程序里面的资源。可以在PyInstaller创建可执行程序时,通过参数 --icon="logo.ico" 指定。比如
xxxxxxxxxxpyinstaller main.py --noconsole --icon="logo.ico"
注意参数一定是存在的ico文件,不能是png等图片文件。如果你只有png文件,可以通过在线的png转ico文件网站,生成ico,比如下面两个网站网站1、网站2
注意:这些应用程序图标ico文件,在使用PyInstaller创建可执行程序时,不需要要拷贝到程序所在目录。因为它已经被嵌入可执行程序了。